Erythropoietin (EPO) is a hormone that stimulates the production of red blood cells in the bone marrow. EPO-stimulating agents have been used to treat anemia in cancer patients, but some research has suggested that this treatment may negatively affect survival. Shedding light on this puzzle, Chiu et al. found that tumor cells require secreted EPO to evade immune attack (see the Perspective by Armouk and Van Ginderachter). Increased tumor-derived EPO levels were associated with an immunosuppressive tumor microenvironment in liver cancer models. Blocking either tumor-secreted EPO or the EPO receptor on macrophages promoted T cell infiltration and improved the control of hepatocellular carcinoma. The EPO/EPO receptor pathway may therefore act as a switch to toggle cancer immunity. —Priscilla N. Kelly
Cancer patients with inflamed (T cell–rich) tumors, indicative of an active antitumor immune response, often benefit from immune checkpoint blockade (ICB) therapy. However, despite the presence of tumor mutations that should theoretically trigger an immune response, most patients have noninflamed (T cell–deprived) tumors and do not benefit from ICB. These noninflamed tumors are typically replete with immunosuppressive macrophages and neutrophils that hinder T cell priming, activation, and homing—critical processes for fostering antitumor immunity. Yet, the mechanisms that determine the immune cell profile or immunotype of tumors remain poorly understood.
To investigate the factors that govern antitumor immunity and tumor immunotype, using somatic gene editing, we developed and compared spontaneous mouse models of hepatocellular carcinoma (HCC) with either an inflamed or a noninflamed tumor microenvironment (TME). Notably, elevated plasma erythropoietin (EPO) and splenomegaly were observed exclusively in noninflamed tumor models, independent of tumor size and without signs of anemia. EPO, a glycoprotein hormone known for stimulating red blood cell production, has recently been implicated in other biological processes involving resolution of inflammation. Clinically, high expression of EPO is linked to poor prognosis in several cancers, including HCC, and accumulation of immunosuppressive cells such as regulatory T cells (Treg cells) and regulatory macrophages. Therefore, we hypothesized that EPO may play a key role in promoting an immunosuppressive, noninflamed TME.
Using both gain-of-function (EPO overexpression in inflamed tumors) and loss-of-function (EPO ablation in noninflamed tumors) approaches, we established that tumor-secreted EPO autonomously shapes the immune landscape of the TME by promoting Treg cells while reducing CD8+ effector memory T cells (TEM cells), thereby facilitating immune evasion. Because we observed that immunoregulatory macrophages constitute the predominant EPO receptor–positive (EPOR+) population in human and mouse HCC, we hypothesized that EPOR+ macrophages directly mediate the effects of EPO and are indispensable for preventing immunosurveillance in HCC. We found that EPOR reduction in macrophages—achieved either through genetic deletion or macrophage-targeted small interfering RNA treatment—significantly inhibited noninflamed tumor growth, even leading to spontaneous regression of some established tumors. This effect was primarily driven by a robust CD8+ TEM cell response, which synergized with ICB. A similar therapeutic synergy was also observed in established noninflamed tumors treated with either tamoxifen-induced EPOR deletion in macrophages or pharmacological inhibition of EPO/EPOR signaling using an EPOR-Fc chimera decoy receptor to neutralize EPO. Mechanistically, we showed that EPO reprograms proinflammatory macrophages—responsible for chemokine production and antigen presentation to recruit and activate effector T cells—into immunoregulatory macrophages with Kupffer cell–like characteristics. Finally, we identified NRF2 (nuclear factor erythroid 2-related factor 2) as a critical downstream mediator of the EPO/EPOR axis, driving heme depletion and antioxidant production in macrophages, which are key events in immunoregulatory macrophage reprogramming.
Using spontaneous preclinical models of HCC with distinct immunotypes, we show that the EPO/EPOR axis functions as an immunosuppressive switch in macrophages that maintains a T cell–deprived TME, thus posing a major barrier to effective antitumor immunity. Inactivation of EPO/EPOR reprograms macrophages to initiate a robust antitumor immune response, converting a noninflamed TME into an inflamed one. These findings point to the EPO/EPOR axis as a promising therapeutic target in HCC. Given that high EPO expression is associated with a poor prognosis across various solid tumors, targeting this axis may prove effective in the treatment of other tumors as well.
Successful cancer immunotherapy requires a patient to mount an effective immune response against tumors; however, many cancers evade the body’s immune system. To investigate the basis for treatment failure, we examined spontaneous mouse models of hepatocellular carcinoma (HCC) with either an inflamed T cell–rich or a noninflamed T cell–deprived tumor microenvironment (TME). Our studies reveal that erythropoietin (EPO) secreted by tumor cells determines tumor immunotype. Tumor-derived EPO autonomously generates a noninflamed TME by interacting with its cognate receptor EPOR on tumor-associated macrophages (TAMs). EPO signaling prompts TAMs to become immunoregulatory through NRF2-mediated heme depletion. Removing either tumor-derived EPO or EPOR on TAMs leads to an inflamed TME and tumor regression independent of genotype, owing to augmented antitumor T cell immunity. Thus, the EPO/EPOR axis functions as an immunosuppressive switch for antitumor immunity.
促红细胞生成素(EPO)是一种刺激骨髓中红细胞生成的激素。EPO刺激剂已被用于治疗癌症患者的贫血,但一些研究表明这种治疗可能会对生存率产生负面影响。Chiu等人阐明了这一谜题,他们发现肿瘤细胞需要分泌的EPO来逃避免疫攻击(见Armouk和Van Ginderachter的观点文章)。在肝癌模型中,肿瘤来源的EPO水平升高与免疫抑制性肿瘤微环境相关。阻断肿瘤分泌的EPO或巨噬细胞上的EPO受体均能促进T细胞浸润并改善肝细胞癌的控制。因此,EPO/EPO受体通路可能充当切换癌症免疫的开关。—Priscilla N. Kelly
具有炎症性(富含T细胞)肿瘤的癌症患者,通常预示着存在活跃的抗肿瘤免疫反应,他们常常能从免疫检查点阻断(ICB)治疗中获益。然而,尽管存在理论上应能触发免疫反应的肿瘤突变,大多数患者仍患有非炎症性(缺乏T细胞)肿瘤,并且无法从ICB中获益。这些非炎症性肿瘤通常充满了免疫抑制性的巨噬细胞和中性粒细胞,它们阻碍了T细胞的启动、激活和归巢——这些都是促进抗肿瘤免疫的关键过程。然而,决定肿瘤免疫细胞特征或免疫类型的机制仍知之甚少。
为了研究调控抗肿瘤免疫和肿瘤免疫类型的因素,我们利用体细胞基因编辑技术,开发并比较了具有炎症性或非炎症性肿瘤微环境(TME)的自发性肝细胞癌(HCC)小鼠模型。值得注意的是,仅在非炎症性肿瘤模型中观察到血浆促红细胞生成素(EPO)水平升高和脾肿大,这与肿瘤大小无关且无贫血迹象。EPO是一种已知能刺激红细胞生成的糖蛋白激素,最近被发现在涉及炎症消退的其他生物学过程中也有作用。临床上,高表达的EPO与包括HCC在内的几种癌症的不良预后相关,并与免疫抑制细胞如调节性T细胞(Treg细胞)和调节性巨噬细胞的积累有关。因此,我们假设EPO可能在促进免疫抑制性、非炎症性TME中起关键作用。
通过功能获得性(在炎症性肿瘤中过表达EPO)和功能缺失性(在非炎症性肿瘤中敲除EPO)两种方法,我们确定肿瘤分泌的EPO通过促进Treg细胞同时减少CD8+效应记忆T细胞(TEM细胞)来自主地塑造TME的免疫景观,从而促进免疫逃逸。由于我们观察到免疫调节性巨噬细胞构成了人和小鼠HCC中主要的EPO受体阳性(EPOR+)群体,我们假设EPOR+巨噬细胞直接介导EPO的效应,并且是防止HCC中免疫监视不可或缺的。我们发现,通过基因缺失或巨噬细胞靶向的小干扰RNA处理实现巨噬细胞中EPOR的减少,能显著抑制非炎症性肿瘤的生长,甚至导致一些已形成肿瘤的自发消退。这种效应主要是由强烈的CD8+ TEM细胞反应驱动的,该反应与ICB协同作用。在使用他莫昔芬诱导的巨噬细胞EPOR缺失或使用EPOR-Fc嵌合诱骗受体中和EPO的药理抑制EPO/EPOR信号通路治疗已形成的非炎症性肿瘤时,也观察到了类似的治疗协同效应。机制上,我们证明EPO将负责产生趋化因子和呈递抗原以募集和激活效应T细胞的促炎性巨噬细胞,重编程为具有库普弗细胞样特征的免疫调节性巨噬细胞。最后,我们确定NRF2(核因子红细胞2相关因子2)是EPO/EPOR轴的一个关键下游介质,驱动巨噬细胞中的血红素耗竭和抗氧化剂产生,这些是免疫调节性巨噬细胞重编程的关键事件。
通过使用具有不同免疫类型的自发性HCC临床前模型,我们证明EPO/EPOR轴在巨噬细胞中充当一种免疫抑制开关,维持一个缺乏T细胞的TME,从而构成了有效抗肿瘤免疫的主要障碍。失活EPO/EPOR可重编程巨噬细胞以启动强烈的抗肿瘤免疫反应,将非炎症性TME转变为炎症性TME。这些发现表明EPO/EPOR轴是HCC中一个有前景的治疗靶点。鉴于高EPO表达与多种实体瘤的不良预后相关,靶向该轴也可能被证明对其他肿瘤的治疗有效。
成功的癌症免疫治疗需要患者对肿瘤产生有效的免疫反应;然而,许多癌症能逃避机体的免疫系统。为了探究治疗失败的基础,我们检查了具有炎症性(富含T细胞)或非炎症性(缺乏T细胞)肿瘤微环境(TME)的自发性肝细胞癌(HCC)小鼠模型。我们的研究揭示,肿瘤细胞分泌的促红细胞生成素(EPO)决定了肿瘤的免疫类型。肿瘤来源的EPO通过与肿瘤相关巨噬细胞(TAMs)上的同源受体EPOR相互作用,自主地生成一个非炎症性TME。EPO信号通过NRF2介导的血红素耗竭促使TAMs转变为免疫调节性。移除肿瘤来源的EPO或TAMs上的EPOR,会由于增强的抗肿瘤T细胞免疫而导致炎症性TME和肿瘤消退,且与基因型无关。因此,EPO/EPOR轴充当了抗肿瘤免疫的免疫抑制开关。
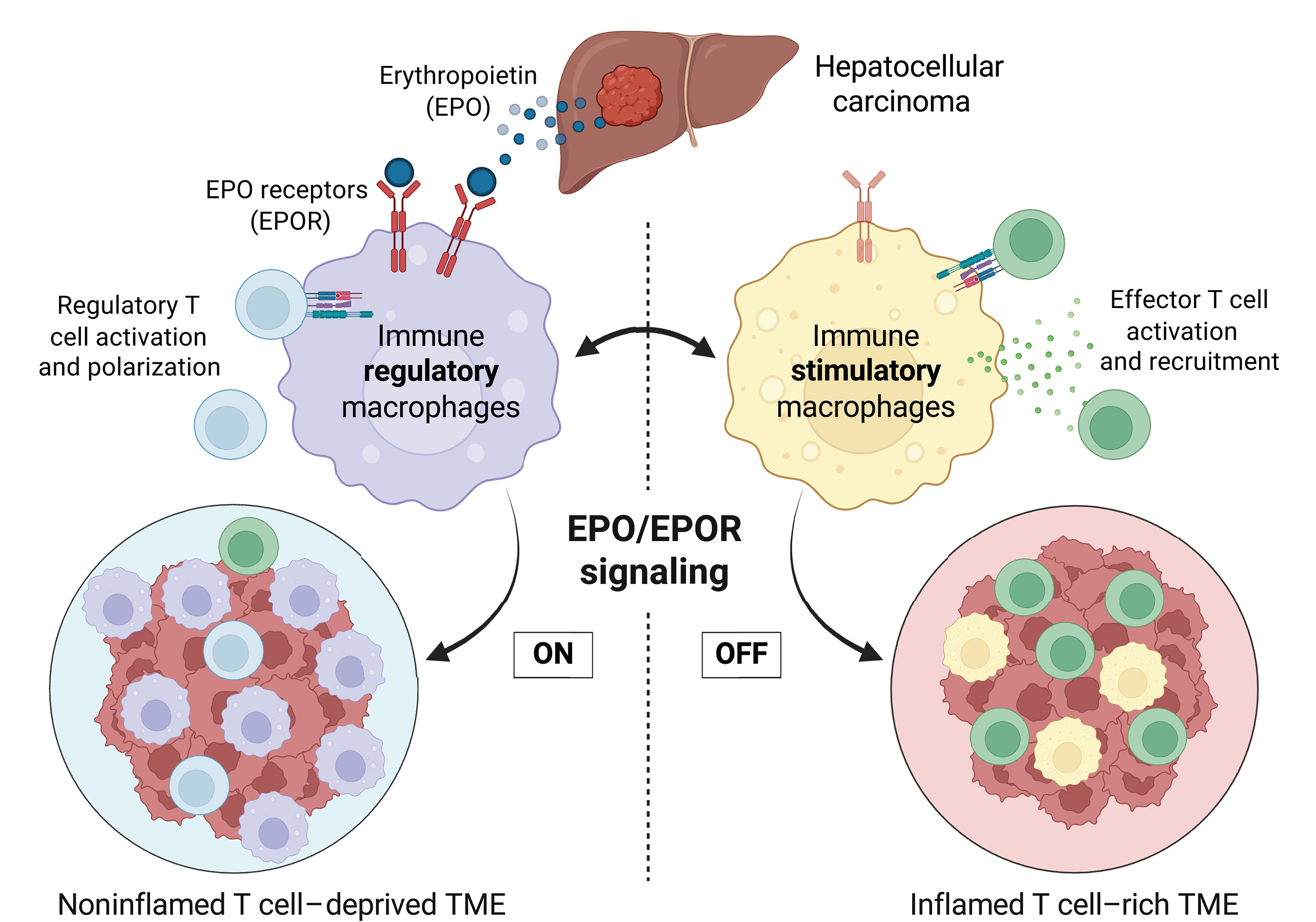
图:EPO/EPOR信号通路在巨噬细胞中塑造了非炎性肿瘤免疫类型。肿瘤来源的EPO与肿瘤相关巨噬细胞上的EPOR结合,阻断了其向免疫刺激型巨噬细胞的分化,而这类细胞原本可驱动效应T细胞(绿色)的活化与募集。相反,EPO刺激下的巨噬细胞呈现免疫调节特性,促进调节性T细胞(蓝色)活化,并维持一种非炎性、T细胞缺失的肿瘤微环境。针对巨噬细胞中EPO/EPOR轴进行干预,可将非炎性肿瘤微环境转化为炎性、免疫活跃的状态。
Tumor-derived erythropoietin acts as an immunosuppressive switch in cancer immunity