Cancer is caused by mutations in DNA that often accompany profound changes to the nucleoprotein structure of the genome (the epigenome) that governs gene expression. Sundaram et al. mapped active regions of regulatory DNA from single cells in diverse human cancer types. Their analysis revealed distinct patterns of open chromatin in different types and genetic subclones of cancer and immune cells in tumors. The data further provide insight into the regulatory phenotypes of breast cancer subtypes and enable the training of cancer-specific neural network models that can interpret the impact of noncoding mutations on the open chromatin landscape. The deep learning models suggest that inherited or cancer-specific mutations can affect important gene-regulatory elements adjacent to, and possibly regulating, key cancer-causing genes. —Priscilla N. Kelly
Genetic lesions are the root cause of cancer and drive gene regulatory changes that evince the classical phenotypic “hallmarks” of cancer. The Cancer Genome Atlas (TCGA) has aimed at understanding the diverse molecular features associated with both these genetic mutations and associated gene regulatory transformations that drive phenotypes across diverse cancer types.
One means of identifying the regulatory elements and trans factors associated with the gene expression has been chromatin accessibility mapping using the assay of transposase-accessible chromatin using sequencing (ATAC-seq), which identifies the fraction of the genome actively bound by regulatory proteins, such as transcription factors (TFs) and polymerases. Although these regulatory elements have been previously explored by TCGA in “bulk” cancer tissue, such analysis convolves malignant signatures from cancer with the landscape of regulatory elements present in associated stromal or immune cells. By contrast, single-cell chromatin accessibility data allows for deconvolution of these signals. Furthermore, a cancer-specific gene regulatory “grammar” can be learned from these cancer-specific data by using deep learning models, allowing both identification of genomic sequences associated with TF binding and the prediction of the effects of cancer-associated mutation on the accessibility of these elements.
We generated a single-cell chromatin accessibility atlas from TCGA archival samples spanning eight cancer types and 74 individual cancer samples composed of 227,063 nuclei. Gene accessibility around marker genes allowed us to identify cancer cells versus tumor-infiltrating stromal and immune cells. Megabase-scale changes in chromatin accessibility signals associated with copy number alteration identified tumor subclones on the basis of differential copy number changes. We observed that tumor subclones can exhibit differential gene regulatory programs associated with TFs affected by subclone-specific copy number alterations.
Using these data, we trained interpretable neural network–based models of cis regulation that nominate specific TF motifs associated with differential chromatin accessibility signals in cancer (as compared with normal tissue) and cancer subtypes. This analysis provides a platform to compare regulatory grammars and regulatory modules from cancerous cells to those of the most similar healthy tissue type. Analysis of breast cancer subtypes demonstrated that the chromatin signature of the basal-like subtype of breast cancer is most like secretory-type luminal epithelial cells, confirming that this molecular subtype is not most epigenomically similar to basal-type normal cells.
We used our neural network models to predict the impact of genetic variation on chromatin accessibility by installing single-nucleotide variants and cancer-specific somatic mutations observed in human cancers or populations into the genome in silico. Mutations predicted to cause either a strong gain or loss of chromatin accessibility were enriched near cancer-associated genes compared with matched, non–cancer-associated gene sets, providing evidence that somatic noncoding mutations can drive changes in cancer-associated chromatin regulation.
The TCGA single-cell atlas of multiple human primary cancer types and interpretable deep learning models for interpreting cis-regulatory elements in cancer comprises new resources for understanding the molecular programs that bring about the malignant phenotypes in primary human cancers.
To identify cancer-associated gene regulatory changes, we generated single-cell chromatin accessibility landscapes across eight tumor types as part of The Cancer Genome Atlas. Tumor chromatin accessibility is strongly influenced by copy number alterations that can be used to identify subclones, yet underlying cis-regulatory landscapes retain cancer type–specific features. Using organ-matched healthy tissues, we identified the “nearest healthy” cell types in diverse cancers, demonstrating that the chromatin signature of basal-like–subtype breast cancer is most similar to secretory-type luminal epithelial cells. Neural network models trained to learn regulatory programs in cancer revealed enrichment of model-prioritized somatic noncoding mutations near cancer-associated genes, suggesting that dispersed, nonrecurrent, noncoding mutations in cancer are functional. Overall, these data and interpretable gene regulatory models for cancer and healthy tissue provide a framework for understanding cancer-specific gene regulation.
肿瘤新抗原是肿瘤细胞表达的蛋白质,可触发消除肿瘤的免疫反应。限制其作为实体瘤(尤其是胰腺癌)成功免疫疗法的一个主要挑战是,此类由大多数肿瘤细胞持续表达的癌症特异性蛋白质和肽段非常罕见。Ely等人对人类胰腺癌进行了免疫肽组学研究,鉴定了大量源自基因组非编码区域的独特肽段(参见Tuveson的展望)。某些肽段具有免疫原性,可作为产生T细胞特异性免疫反应的来源,在临床前模型中能够杀伤胰腺癌。部分源自癌症的肽段为多名患者所共有,且正常胰腺细胞中不存在,这提升了通用“现货型”疗法的前景,相较于更具挑战性的个性化方法而言。—Priscilla N. Kelly
癌症由DNA突变引起,这些突变通常伴随着控制基因表达的基因组核蛋白结构(表观基因组)的深刻变化。Sundaram等人从不同人类癌症类型的单细胞中绘制了调控DNA的活性区域。他们的分析揭示了肿瘤中不同类型和遗传亚克隆的癌细胞及免疫细胞之间存在不同的开放染色质模式。这些数据进一步提供了对乳腺癌亚型调控表型的见解,并能够训练癌症特异性神经网络模型,以解释非编码突变对开放染色质景观的影响。深度学习模型表明,遗传性或癌症特异性突变可能影响邻近关键致癌基因(并可能调控这些基因)的重要基因调控元件。—Priscilla N. Kelly
遗传病变是癌症的根本原因,并驱动基因调控变化,从而呈现出经典的癌症表型"特征"。癌症基因组图谱计划旨在理解与这些基因突变以及驱动不同癌症类型表型的相关基因调控转变相关的多样化分子特征。
识别与基因表达相关的调控元件和反式因子的一种方法是利用转座酶可及染色质测序技术进行染色质可及性作图,该技术可识别基因组中被调控蛋白(如转录因子和聚合酶)活跃结合的部分。尽管癌症基因组图谱计划先前已在"批量"癌症组织中探索过这些调控元件,但此类分析将来自癌症的恶性特征与相关基质或免疫细胞中存在的调控元件景观混合在一起。相比之下,单细胞染色质可及性数据允许对这些信号进行解卷积。此外,通过使用深度学习模型,可以从这些癌症特异性数据中学习到癌症特异性的基因调控"语法",从而既能识别与转录因子结合相关的基因组序列,又能预测癌症相关突变对这些元件可及性的影响。
我们利用来自癌症基因组图谱计划的存档样本,生成了一个单细胞染色质可及性图谱,涵盖八种癌症类型和74个独立的癌症样本,共包含227,063个细胞核。标记基因周围的基因可及性使我们能够区分癌细胞与肿瘤浸润的基质和免疫细胞。与拷贝数改变相关的染色质可及性信号的兆碱基规模变化,基于差异拷贝数变化识别出了肿瘤亚克隆。我们观察到,肿瘤亚克隆可以表现出与受亚克隆特异性拷贝数改变影响的转录因子相关的差异基因调控程序。
利用这些数据,我们训练了基于可解释神经网络的顺式调控模型,这些模型能够提名与癌症(与正常组织相比)及癌症亚型中差异染色质可及性信号相关的特定转录因子基序。该分析提供了一个平台,用于将癌细胞的调控语法和调控模块与最相似的健康组织类型进行比较。对乳腺癌亚型的分析表明,基底样亚型乳腺癌的染色质特征最类似于分泌型腔上皮细胞,证实了这种分子亚型在表观基因组上与基底型正常细胞最不相似。
我们使用我们的神经网络模型,通过在计算机模拟中将人类癌症或人群中观察到的单核苷酸变异和癌症特异性体细胞突变插入基因组,来预测遗传变异对染色质可及性的影响。与匹配的非癌症相关基因集相比,被预测会导致染色质可及性强烈增加或减少的突变在癌症相关基因附近富集,这为体细胞非编码突变可以驱动癌症相关染色质调控变化提供了证据。
涵盖多种人类原发性癌症类型的癌症基因组图谱计划单细胞图谱,以及用于解释癌症中顺式调控元件的可解释深度学习模型,共同构成了理解导致人类原发性癌症恶性表型的分子程序的新资源。
为了识别癌症相关的基因调控变化,我们作为癌症基因组图谱计划的一部分,生成了跨越八种肿瘤类型的单细胞染色质可及性图谱。肿瘤染色质可及性受到拷贝数改变的强烈影响,这些改变可用于识别亚克隆,但潜在的顺式调控景观保留了癌症类型特异性特征。利用器官匹配的健康组织,我们在多种癌症中识别了"最接近的健康"细胞类型,证明基底样亚型乳腺癌的染色质特征最类似于分泌型腔上皮细胞。经训练用于学习癌症调控程序的神经网络模型显示,模型优先考虑的体细胞非编码突变在癌症相关基因附近富集,这表明癌症中分散、非复发、非编码的突变是具有功能的。总体而言,这些数据以及针对癌症和健康组织的可解释基因调控模型,为理解癌症特异性基因调控提供了一个框架。
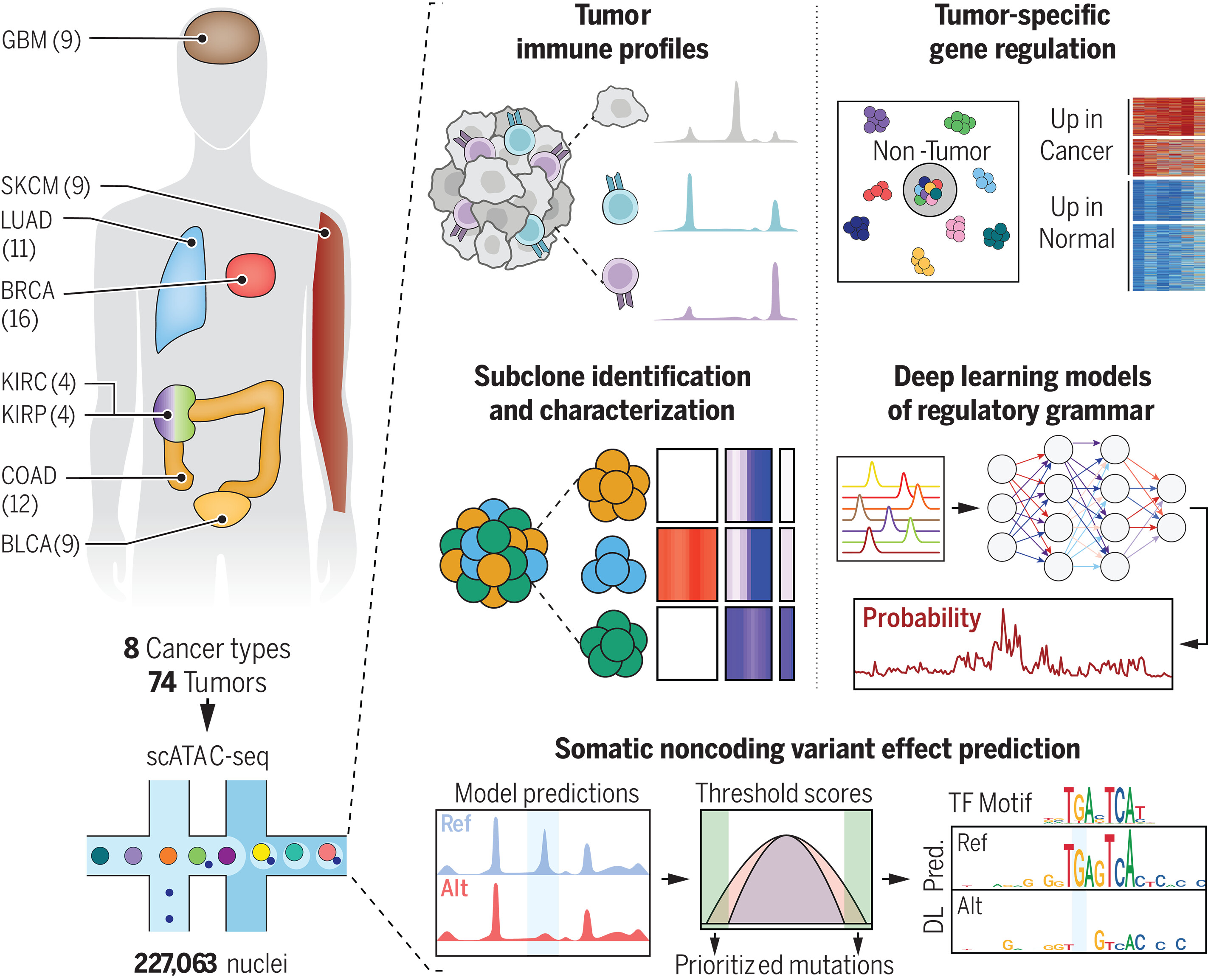
图:一项泛癌种单细胞染色质可及性基因调控图谱。该图谱基于基因规模可及性模式区分肿瘤细胞与其他细胞类型,并通过兆碱基规模的变化实现肿瘤亚克隆异质性分析。神经网络模型揭示了癌症相对于健康组织特有的染色质可及性特征,并强调了非编码区体细胞突变在癌症中的作用。scATAC-seq:单细胞ATAC测序;DL Pred.:深度学习预测;Ref:参考序列;Alt:替代序列。
Single-cell chromatin accessibility reveals malignant regulatory programs in primary human cancers