Mutations in the KRAS gene are one of the most frequent oncogenic events in human cancer. Drugs that inhibit KRAS have recently been approved for the treatment of KRAS-mutant tumors, but their clinical efficacy is limited by primary innate mechanisms and by treatment-associated resistance. To better understand how KRAS-driven tumors grow and resist therapy, J. A. Klomp et al. established a KRAS-regulated gene transcriptome in KRAS-mutant pancreatic cancer. The KRAS mutant transcriptome was found to be regulated largely through activation of the ERK mitogen-activated protein kinase cascade. In a separate study, J. E. Klomp et al. compiled a comprehensive molecular portrait of aberrant ERK signaling in KRAS-mutated pancreatic cancer and identified more than 1500 ERK substrates. These studies advance our understanding of how ERK supports KRAS-dependent cancer growth and may inform next-generation therapies using KRAS and ERK inhibitors. —Priscilla N. Kelly
Mutationally activated KRAS causes persistent activation of the RAF-MEK-ERK mitogen-activated protein kinase (MAPK) cascade. Whereas RAF and MEK kinases phosphorylate a highly restricted set of substrates, the closely related ERK1 and ERK2 serine/ threonine kinases directly or indirectly phosphorylate a diversity of functionally distinct proteins. Analyses of the “logical suspects” have validated the roles of specific ERK substrates (e.g., MYC and FRA1), yet a systemwide determination of ERK substrates that facilitate mutant KRAS-driven oncogenesis remains to be completed.
Nearly four decades after the discovery of KRAS mutations in cancer, clinically effective inhibitors of KRAS have been approved for KRASG12C-mutant non–small-cell lung cancer. However, only <50% of patients respond initially, and most responsive patients become resistant and relapse within 6 months. DNA sequencing of tumors and circulating tumor DNA (ctDNA) from relapsed patients identified mutational activation (e.g., of RAF or MEK) or inactivation (e.g., NF1) of components within the RAS signaling network that result in reactivation of ERK MAPK signaling. Although numerous mass spectrometry analyses have collectively identified >1300 ERK1/2 direct or indirect substrates, the limited overlap among different studies suggests that the full spectrum of ERK-dependent phosphorylation events has not been elucidated. In this study, we applied mass spectrometry using improved methodology and pooled data from a panel of heterogeneous cancer cell lines to generate a system-wide determination of the ERK-dependent phosphoprotein signaling network in the most KRAS-addicted cancer, pancreatic ductal adenocarcinoma (PDAC).
Previous phosphoproteomic studies to catalog ERK substrates used a diversity of cell types, involved different ERK activation stimuli, and applied varied experimental designs. We focused on pathologic ERK signaling driven by endogenous oncogenic KRAS in PDAC. We first developed constitutively activated variants of ERK1 and ERK2 and showed that they play redundant roles. Each alone is sufficient to support mutant KRAS–dependent PDAC growth, and they stimulate nearly identical transcriptional signaling outputs. Next, evaluating phosphorylation changes caused by pharmacologic inhibition of ERK, we captured immediate direct activities (1 hour) and delayed secondary activities (24 hours) induced by loss of chronic ERK activation. We identified 2123 ERK-dependent phosphoproteins, 67% of which were not previously associated with ERK, thus greatly expanding the depth and breadth of the ERK-regulated phosphoproteome. We then applied our recently annotated serine/threonine kinome motif database to establish the protein kinase signaling network that drives the ERK-dependent phosphoproteome. At 1 hour, loss of ERK- and RSK (a direct ERK substrate)–dependent phosphorylation predominated. By contrast, at 24 hours, loss of cyclin-dependent kinase (CDK1-6)–dependent phosphorylation predominated, accompanied by activation of other kinases, indicating dynamic alterations over time. Integrating pathway analyses and information from the Cancer Dependency Map portal (DepMap), we showed that 17% of the ERK phosphoproteome is essential for PDAC growth, and it is enriched in nuclear-localized proteins and in proteins involved in cell cycle regulation and RHO guanosine triphosphatase (GTPase) signaling. The PDAC-derived KRAS-dependent phosphoproteome is driven nearly exclusively through ERK and is applicable to other tissues and species.
This study greatly expanded the depth and breadth of ERK-regulated direct and indirect phosphoproteins. Given that reactivation of ERK is a major basis for acquired resistance to KRAS inhibitor therapy, this comprehensive molecular portrait of mutant KRAS–driven pathologic ERK signaling will be important to elucidate the mechanisms of patient response and resistance to KRAS inhibitors.
To delineate the mechanisms by which the ERK1 and ERK2 mitogen-activated protein kinases support mutant KRAS–driven cancer growth, we determined the ERK-dependent phosphoproteome in KRAS-mutant pancreatic cancer. We determined that ERK1 and ERK2 share near-identical signaling and transforming outputs and that the KRAS-regulated phosphoproteome is driven nearly completely by ERK. We identified 4666 ERK-dependent phosphosites on 2123 proteins, of which 79 and 66%, respectively, were not previously associated with ERK, substantially expanding the depth and breadth of ERK-dependent phosphorylation events and revealing a considerably more complex function for ERK in cancer. We established that ERK controls a highly dynamic and complex phosphoproteome that converges on cyclin-dependent kinase regulation and RAS homolog guanosine triphosphatase function (RHO GTPase). Our findings establish the most comprehensive molecular portrait and mechanisms by which ERK drives KRAS-dependent pancreatic cancer growth.
KRAS基因突变是人类癌症中最常见的致癌事件之一。抑制KRAS的药物最近已被批准用于治疗KRAS突变肿瘤,但其临床疗效受到原发性固有机制和治疗相关耐药性的限制。为了更好地理解KRAS驱动肿瘤如何生长及抵抗治疗,J. A. Klomp 等人建立了KRAS突变胰腺癌中受KRAS调控的基因转录组。研究发现KRAS突变转录组主要通过ERK丝裂原活化蛋白激酶级联的激活来调控。在另一项独立研究中,J. E. Klomp 等人汇编了KRAS突变胰腺癌中异常ERK信号传导的全面分子图谱,并识别出超过1500个ERK底物。这些研究增进了我们对ERK如何支持KRAS依赖性癌症生长的理解,并可能为使用KRAS和ERK抑制剂的下一代疗法提供信息。—Priscilla N. Kelly
突变激活的KRAS导致RAF-MEK-ERK丝裂原活化蛋白激酶级联的持续激活。尽管RAF和MEK激酶磷酸化一组高度受限的底物,但密切相关的ERK1和ERK2丝氨酸/苏氨酸激酶直接或间接磷酸化多种功能不同的蛋白质。对“逻辑嫌疑分子”的分析验证了特定ERK底物的作用,但对于促进突变KRAS驱动肿瘤发生的ERK底物进行系统范围的确定仍有待完成。
在发现KRAS突变于癌症中近四十年后,临床有效的KRAS抑制剂已被批准用于治疗KRASG12C突变的非小细胞肺癌。然而,仅有<50%的患者初始有反应,并且大多数有反应的患者会在6个月内产生耐药并复发。对复发患者的肿瘤和循环肿瘤DNA进行测序,发现了RAS信号网络内部元件的突变激活或失活,这导致ERK MAPK信号传导的重新激活。尽管大量质谱分析共同识别了>1300个ERK1/2直接或间接底物,但不同研究之间有限的重叠表明,尚未阐明ERK依赖性磷酸化事件的完整谱系。在本研究中,我们应用改进方法的质谱分析,并汇总了一组异质性癌细胞系的数据,以在最依赖KRAS的癌症——胰腺导管腺癌中,生成对ERK依赖性磷酸化蛋白质信号网络的系统范围确定。
先前用于编录ERK底物的磷酸化蛋白质组学研究使用了多种细胞类型、涉及不同的ERK激活刺激,并应用了多样的实验设计。我们专注于PDAC中由内源性致癌KRAS驱动的病理性ERK信号传导。我们首先构建了ERK1和ERK2的持续激活变体,并证明它们扮演冗余的角色。任一单独变体都足以支持突变KRAS依赖的PDAC生长,并且它们刺激几乎相同的转录信号输出。接下来,通过评估由ERK药理学抑制引起的磷酸化变化,我们捕获了由慢性ERK激活缺失诱导的即时直接活性以及延迟的次级活性。我们识别了2123个ERK依赖性磷酸化蛋白质,其中67%此前与ERK无关,从而极大地扩展了ERK调控的磷酸化蛋白质组的深度和广度。然后,我们应用最近注释的丝氨酸/苏氨酸激酶组基序数据库,建立了驱动ERK依赖性磷酸化蛋白质组的蛋白激酶信号网络。在1小时时,ERK和RSK依赖性磷酸化的缺失占主导。相比之下,在24小时时,细胞周期蛋白依赖性激酶依赖性磷酸化的缺失占主导,并伴随其他激酶的激活,表明其随时间发生动态变化。通过整合通路分析和来自癌症依赖性图谱门户的信息,我们表明17%的ERK磷酸化蛋白质组对PDAC生长至关重要,并且它在核定位蛋白以及参与细胞周期调控和RHO鸟苷三磷酸酶信号传导的蛋白质中富集。源自PDAC的KRAS依赖性磷酸化蛋白质组几乎完全通过ERK驱动,并且适用于其他组织和物种。
这项研究极大地扩展了ERK调控的直接和间接磷酸化蛋白质的深度和广度。鉴于ERK的重新激活是获得性耐药于KRAS抑制剂治疗的主要基础,这份突变KRAS驱动的病理性ERK信号传导的全面分子图谱,对于阐明患者对KRAS抑制剂产生反应和耐药的机制至关重要。
为阐明ERK1和ERK2这两种丝裂原活化蛋白激酶支持突变KRAS驱动癌症生长的机制,我们测定了KRAS突变型胰腺癌中ERK依赖性磷酸化蛋白质组。研究发现,ERK1与ERK2具有高度一致的信号传导和转化功能,且KRAS调控的磷酸化蛋白质组几乎完全由ERK驱动。我们在2123种蛋白质上鉴定出4666个ERK依赖性磷酸化位点,其中分别有79%和66%的位点此前未发现与ERK相关,这显著拓展了ERK依赖性磷酸化事件的深度与广度,揭示了ERK在癌症中更为复杂的功能。我们证实ERK调控着一个高度动态且复杂的磷酸化蛋白质组网络,该网络聚焦于细胞周期蛋白依赖性激酶调控及RAS同源鸟苷三磷酸酶功能(RHO GTP酶)。本研究建立了最全面的分子图谱与机制体系,阐释了ERK如何驱动KRAS依赖性胰腺癌的生长。
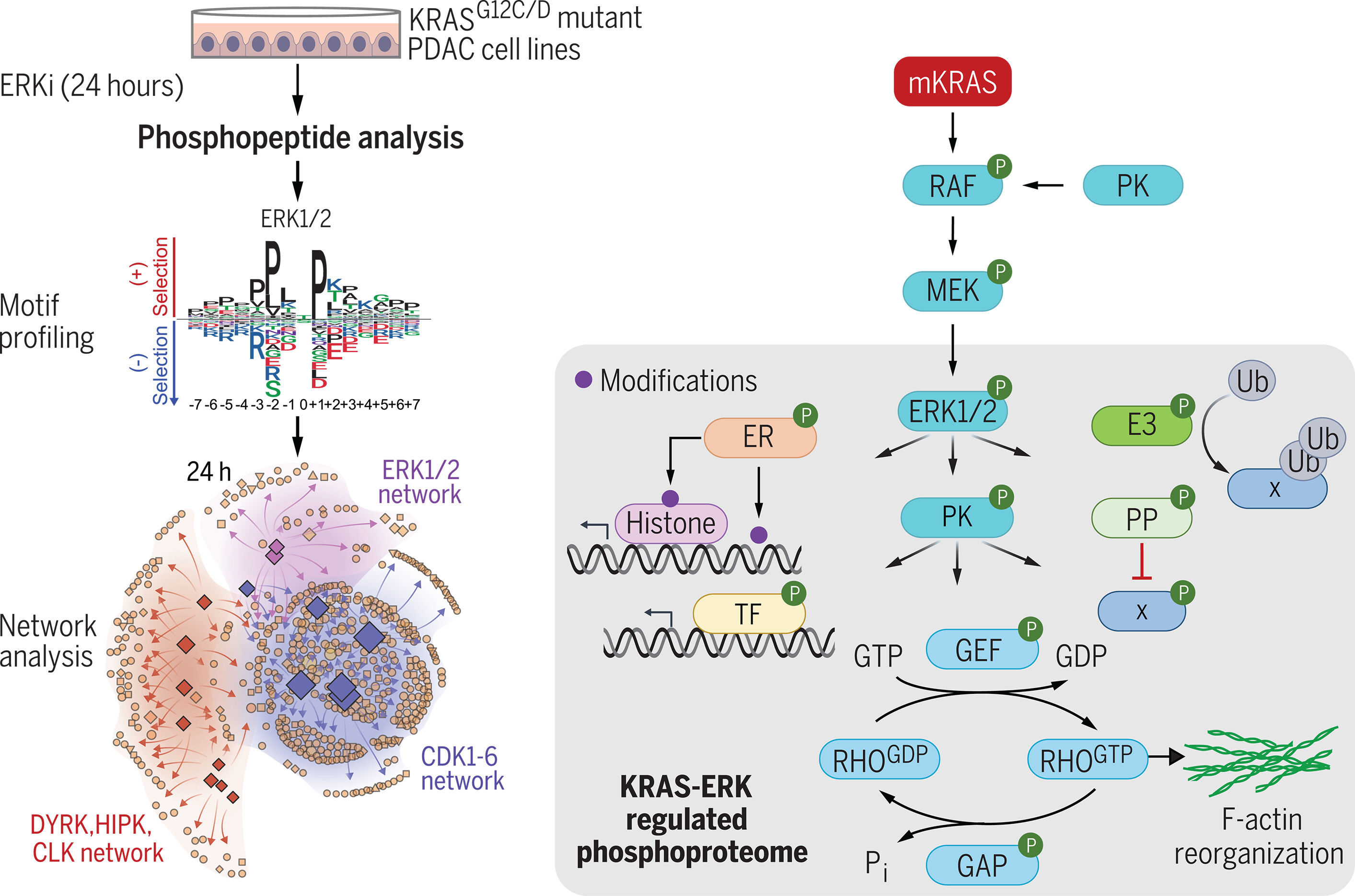
图:ERK调控一个复杂的磷酸化蛋白质组。 磷酸化蛋白质组学分析确立了ERK调控的磷酸化蛋白质组。激酶组基序分析揭示了驱动ERK调控磷酸化的激酶网络。ERK底物包括扩展ERK调控磷酸化蛋白质组的蛋白激酶(PK)和磷酸酶(PP),以及驱动ERK依赖性转录组的转录因子(TF)和表观遗传调控因子(ER)。ERK信号的主要输出调控细胞周期(CDKs)和RHO GTPases。ERKi,ERK1/2选择性抑制剂SCH772984;HIPK,同源结构域相互作用蛋白激酶;CLK,CDK样蛋白激酶;DYRK,双特异性酪氨酸磷酸化调节激酶;GDP,二磷酸鸟苷;GTP,三磷酸鸟苷;mKRAS,突变型KRAS;GEF,鸟苷酸交换因子;GAP,GTP酶激活蛋白;Pi,无机磷酸盐;Ub,泛素。
Determining the ERK-regulated phosphoproteome driving KRAS-mutant cancer