Mutations in the KRAS gene are one of the most frequent oncogenic events in human cancer. Drugs that inhibit KRAS have recently been approved for the treatment of KRAS-mutant tumors, but their clinical efficacy is limited by primary innate mechanisms and by treatment-associated resistance. To better understand how KRAS-driven tumors grow and resist therapy, J. A. Klomp et al. established a KRAS-regulated gene transcriptome in KRAS-mutant pancreatic cancer. The KRAS mutant transcriptome was found to be regulated largely through activation of the ERK mitogen-activated protein kinase cascade. In a separate study, J. E. Klomp et al. compiled a comprehensive molecular portrait of aberrant ERK signaling in KRAS-mutated pancreatic cancer and identified more than 1500 ERK substrates. These studies advance our understanding of how ERK supports KRAS-dependent cancer growth and may inform next-generation therapies using KRAS and ERK inhibitors. —Priscilla N. Kelly
Recent US Food and Drug Administration approval of direct inhibitors of G12C (Gly12→Cys) mutations in the formerly “undruggable” KRAS marks an important milestone in cancer drug discovery, and inhibitors of more prevalent KRAS mutations [G12D/V (Gly12→Asp or Val), and others] are now in clinical evaluation. However, notably few patients respond initially, and most of those individuals relapse quickly. Defining genetic markers and drivers of primary and treatment-associated acquired resistance to KRAS inhibitors will be essential to achieve broader and more durable responses.
A major molecular output of aberrant KRAS activation involves systemwide deregulation of gene transcription. Despite numerous efforts to establish KRAS-associated gene transcription signatures, present signatures show notably limited overlap, likely reflecting divergent experimental strategies and cancer models. In this work, we sought to define a comprehensive KRAS-dependent transcriptional signature that detects target inhibition in KRAS-mutant cancer patients treated with KRAS mutation–selective inhibitors.
Most of the previous KRAS signatures, including the present gold-standard Hallmark KRAS signaling gene sets, profiled gene expression changes caused by persistent steady-state expression of mutant KRAS. Instead, we applied RNA sequencing (RNA-seq) to determine transcriptional changes caused by acute KRAS suppression in endogenously KRAS-mutant pancreatic ductal adenocarcinoma (PDAC) cell lines, thereby limiting the confounding effects of compensation for the loss of KRAS signaling. In contrast to the Hallmark, our KRAS gene signature was strongly enriched in changes in response to pharmacologic inhibition of mutant KRAS in KRAS-mutant PDAC cell lines and tumors, as well as in lung and colorectal xenograft tumors. Thus, the KRAS-regulated transcriptome may be broadly applicable. Despite the plethora of validated and putative KRAS effectors, we found that RAF-MEK-ERK mitogen-activated protein kinase (MAPK) effector signaling alone, but not the PI3K-AKT-mTORC1 pathway, showed that sufficient to support mutant KRAS-dependent PDAC growth. Consistent with this, the KRAS-regulated transcriptome was driven largely through ERK MAPK activity. Pathway analyses showed that our merged KRAS- and ERK-dependent gene signature composed of 278 up-regulated genes was highly enriched in cell cycle processes. A comparison of the ERK-regulated transcriptome and total proteome showed that ~80% of the regulation of protein expression changes was at the level of gene transcription. Another subset was at the level of posttranscriptional mechanisms, including ERK phosphorylation and modulation of the anaphase promoting complex/cyclosome (APC/C), which is involved in cell cycle regulation. Finally, our KRAS-ERK gene signature accurately detected KRAS-ERK target inhibition, and that inhibition correlated generally with clinical responses in KRAS-mutant cancer patients treated with KRAS or ERK inhibitors. An accurate portrait of the molecular output that mirrors aberrant KRAS signaling in cancer patients will further elucidate mechanistically how KRAS drives cancer.
We established that an aged immune system promotes tumor growth, regardless of the age of the tumor or its surrounding stroma. Specifically, hematopoietic aging drives emergency myelopoiesis, and targeting IL-1R1 signaling during early tumor development to attenuate this process abrogates the protumorigenic effect of aging on tumor control. Our study not only highlights the importance of immunotherapy in improving the age-dependent antitumor response but also the major role of local myeloid progenitors that actually fuel age-enhanced emergency myelopoiesis in a positive feedback loop. Notably, we defined the therapeutic window for the use of an IL-1R1–based intervention to delay lung cancer progression. Our findings have direct relevance to the design of cancer prevention strategies, support recent work from our group on lung cancer as a systemic disease involving the bone marrow, and are highly informative for unraveling the link between aging, DNMT3A mutation-driven clonal hematopoiesis, and cancer.
How the KRAS oncogene drives cancer growth remains poorly understood. Therefore, we established a systemwide portrait of KRAS- and extracellular signal–regulated kinase (ERK)–dependent gene transcription in KRAS-mutant cancer to delineate the molecular mechanisms of growth and of inhibitor resistance. Unexpectedly, our KRAS-dependent gene signature diverges substantially from the frequently cited Hallmark KRAS signaling gene signature, is driven predominantly through the ERK mitogen-activated protein kinase (MAPK) cascade, and accurately reflects KRAS- and ERK-regulated gene transcription in KRAS-mutant cancer patients. Integration with our ERK-regulated phospho- and total proteome highlights ERK deregulation of the anaphase promoting complex/cyclosome (APC/C) and other components of the cell cycle machinery as key processes that drive pancreatic ductal adenocarcinoma (PDAC) growth. Our findings elucidate mechanistically the critical role of ERK in driving KRAS-mutant tumor growth and in resistance to KRAS-ERK MAPK targeted therapies.
KRAS基因突变是人类癌症中最常见的致癌事件之一。抑制KRAS的药物最近已被批准用于治疗KRAS突变肿瘤,但其临床疗效受到原发性先天机制和治疗相关耐药性的限制。为了更好地理解KRAS驱动肿瘤如何生长和抵抗治疗,J. A. Klomp 等人建立了KRAS突变胰腺癌中受KRAS调控的基因转录组。研究发现,KRAS突变转录组主要通过ERK丝裂原活化蛋白激酶级联反应的激活来调控。在另一项独立研究中,J. E. Klomp 等人汇编了KRAS突变胰腺癌中异常ERK信号传导的综合分子图谱,并鉴定了1500多个ERK底物。这些研究增进了我们对ERK如何支持KRAS依赖性癌症生长的理解,并可能为使用KRAS和ERK抑制剂的下一代疗法提供信息。—Priscilla N. Kelly
美国食品药品监督管理局最近批准了针对先前“不可成药”的KRAS中G12C(甘氨酸12→半胱氨酸)突变的直接抑制剂,这标志着癌症药物发现的一个重要里程碑,针对更普遍KRAS突变[G12D/V(甘氨酸12→天冬氨酸或缬氨酸)等]的抑制剂目前也正处于临床评估阶段。然而,值得注意的是,最初有反应的患者很少,并且这些患者中的大多数会迅速复发。确定对KRAS抑制剂的原发性和治疗相关获得性耐药的遗传标记和驱动因素,对于实现更广泛和更持久的治疗反应至关重要。
异常KRAS激活的一个主要分子输出涉及基因转录的全系统失调。尽管为建立KRAS相关基因转录特征付出了诸多努力,但现有的特征显示出明显的有限重叠,这可能反映了不同的实验策略和癌症模型。在这项工作中,我们试图定义一个全面的KRAS依赖性转录特征,用于检测接受KRAS突变选择性抑制剂治疗的KRAS突变癌症患者中的靶点抑制情况。
先前大多数KRAS特征,包括当前金标准的Hallmark KRAS信号基因集,分析的是由突变KRAS持续稳定表达引起的基因表达变化。相反,我们应用RNA测序来确定在内源性KRAS突变的胰腺导管腺癌细胞系中,由急性KRAS抑制引起的转录变化,从而限制了因补偿KRAS信号丢失而产生的混杂效应。与Hallmark特征相比,我们的KRAS基因特征在KRAS突变PDAC细胞系和肿瘤、以及肺癌和结直肠癌异种移植瘤中,对突变KRAS的药理学抑制反应的变化方面表现出强烈的富集。因此,KRAS调控的转录组可能具有广泛的适用性。尽管存在大量已验证和推定的KRAS效应因子,但我们发现,仅有RAF-MEK-ERK丝裂原活化蛋白激酶效应因子信号传导(而非PI3K-AKT-mTORC1通路)足以支持突变KRAS依赖性的PDAC生长。与此一致的是,KRAS调控的转录组主要由ERK MAPK活性驱动。通路分析显示,我们合并的由278个上调基因组成的KRAS和ERK依赖性基因特征在细胞周期过程中高度富集。对ERK调控的转录组和总蛋白质组的比较表明,约80%的蛋白质表达变化调控发生在基因转录水平。另一部分发生在转录后机制水平,包括ERK磷酸化以及参与细胞周期调控的后期促进复合物/环体的调节。最后,我们的KRAS-ERK基因特征能准确检测KRAS-ERK靶点抑制,并且这种抑制通常与接受KRAS或ERK抑制剂治疗的KRAS突变癌症患者的临床反应相关。一幅准确反映癌症患者中异常KRAS信号传导的分子输出图谱,将进一步从机制上阐明KRAS如何驱动癌症。
我们证实,衰老的免疫系统会促进肿瘤生长,而与肿瘤或其周围基质的年龄无关。具体而言,造血老化驱动应急性骨髓生成,而在肿瘤早期发展阶段靶向IL-1R1信号传导以减弱此过程,可以消除衰老对肿瘤控制的促肿瘤效应。我们的研究不仅强调了免疫疗法在改善年龄依赖性抗肿瘤反应中的重要性,也强调了实际上通过正反馈循环助长年龄增强的应急性骨髓生成的局部骨髓祖细胞的主要作用。值得注意的是,我们确定了使用基于IL-1R1的干预措施来延缓肺癌进展的治疗窗口。我们的发现与癌症预防策略的设计直接相关,支持了我们课题组最近关于肺癌作为一种涉及骨髓的系统性疾病的研究,并且对于揭示衰老、DNMT3A突变驱动的克隆性造血和癌症之间的联系具有很高的参考价值。
KRAS致癌基因如何驱动癌症生长仍然知之甚少。因此,我们在KRAS突变癌症中建立了KRAS和细胞外信号调节激酶依赖性基因转录的全系统图谱,以阐明生长和抑制剂耐药的分子机制。出乎意料的是,我们的KRAS依赖性基因特征与经常被引用的Hallmark KRAS信号基因特征存在显著差异,主要通过ERK丝裂原活化蛋白激酶级联反应驱动,并准确反映了KRAS突变癌症患者中KRAS和ERK调控的基因转录。与我们的ERK调控的磷酸化及总蛋白质组的整合分析,突出了ERK对后期促进复合物/环体以及其他细胞周期机制组分的失调是驱动胰腺导管腺癌生长的关键过程。我们的研究从机制上阐明了ERK在驱动KRAS突变肿瘤生长以及对KRAS-ERK MAPK靶向疗法产生耐药性中的关键作用。
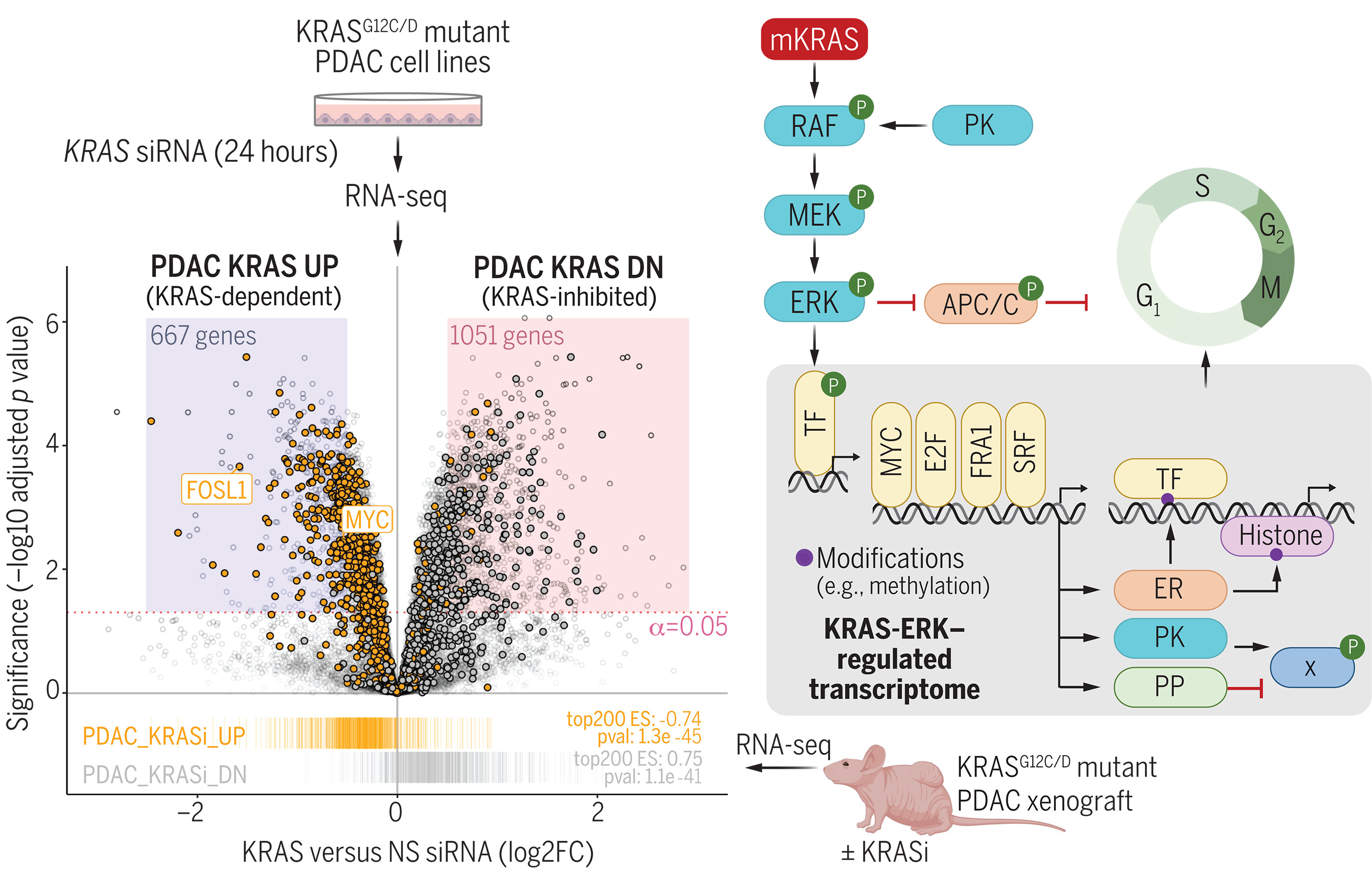
图:KRAS通过ERK依赖性机制导致基因转录的全系统性失调。ERK MAPK效应信号网络调控APC/C细胞周期调节复合体的活性,以及一系列功能各异的蛋白质,包括转录因子(TF)癌蛋白[如MYC和FRA1(FOSL1)]。ERK调控的基因编码额外的转录因子和表观遗传调节因子(ER),这些调节因子可修饰组蛋白和DNA,并编码蛋白激酶(PK)与磷酸酶(PP),从而调控基因转录和蛋白质磷酸化的次级变化。E2F:E2启动子结合因子;G12C/D:甘氨酸12→半胱氨酸或天冬氨酸;KRASi:KRAS抑制剂;NS:非特异性;siRNA:小干扰RNA;SRF:血清反应因子;X:其他底物。[插图部分使用BioRender.com制作]
Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers