Aneuploidies, which are changes in the numbers of whole chromosomes or chromosome arms, are common in cancer, but their contributions to cancer cell survival have been difficult to pinpoint. Girish et al. developed a chromosome-engineering tool to orchestrate the targeted loss of aneuploid chromosome arms and thereby compare isogenic cancer cell lines with and without selected trisomies. The authors discovered that trisomy of chromosome 1q in particular is advantageous to cancer cells and phenocopies the loss of tumor suppressor TP53 signaling. Tumors with this aneuploidy are sensitive to compounds activated by an enzyme encoded on chromosome 1q, suggesting a potential therapeutic approach. —Yevgeniya Nusinovich
It has been known for more than 100 years that human cancers exhibit pervasive aneuploidy, or chromosome copy number changes. For instance, about 25% of cancers exhibit gains of the q arm of chromosome 1. However, despite the prevalence of aneuploidy across cancer types, its role in tumorigenesis has remained poorly defined. Our ability to uncover the function of these large-scale copy number alterations has been hampered by our inability to experimentally manipulate chromosome dosage in cancer. Nonetheless, as aneuploidy is common across malignancies but rare in normal tissue, drugs that exhibit selective toxicity toward aneuploid cells could be useful anticancer agents.
Although aneuploidies have resisted close analysis, previous research has led to the discovery of a phenomenon called “oncogene addiction.” An oncogene-addicted cancer is dependent on the expression of an individual oncogene for continued malignant growth, and loss or inhibition of that oncogene is sufficient to induce cancer regression. As specific aneuploidies such as the gain of chromosome 1q are frequent events in diverse cancer types, we hypothesized that certain aneuploidies could themselves represent oncogene-like cancer addictions. To test this hypothesis, we developed ReDACT (Restoring Disomy in Aneuploid cells using CRISPR Targeting), a set of chromosome engineering tools that allow us to eliminate individual aneuploid chromosomes from cancer genomes. Using ReDACT, we created and then characterized a panel of isogenic cells that have or lack common cancer aneuploidies.
We found that eliminating the trisomy of chromosome 1q from cancer cell lines harboring this alteration almost completely abolished anchorage-independent growth and xenograft formation. Similarly, eliminating the 1q trisomy from a nonmalignant cell line blocked RAS-mediated transformation. Prolonged growth in vitro or in vivo after aneuploidy elimination in cancer cell lines led to karyotype evolution, and 1q-disomic cells were eventually outcompeted by cells that had recovered the 1q trisomy. In contrast, removing other trisomic chromosomes from cancer cells had variable effects on malignant growth, demonstrating that different aneuploidies have distinct phenotypic consequences for cancer development.
An analysis of clinical sequencing data demonstrated that chromosome 1q gains arise early during tumorigenesis and are mutually exclusive with mutations in the tumor suppressor TP53, suggesting that 1q trisomies could represent a mutation-independent mechanism for blocking p53 signaling. Consistent with this, we demonstrated that ReDACT-mediated elimination of chromosome 1q trisomies increased the expression of p53 target genes in TP53 wild-type cell lines. We traced this suppression of p53 function to the triplication of MDM4, a p53 inhibitor encoded on chromosome 1q, and we found that deleting a single copy of MDM4 impaired the growth of 1q-trisomic cells, whereas moderate overexpression of MDM4 rescued the growth of 1q-disomic cells.
Finally, we demonstrated that chromosome 1q gains result in the overexpression of UCK2, a nucleotide kinase encoded on chromosome 1q that is also required for the cytotoxicity of certain anticancer nucleotide analogs. We determined that several different 1q-trisomic cell lines displayed enhanced sensitivity to these compounds owing to the up-regulation of UCK2, revealing that 1q aneuploidy can also represent a tractable cancer vulnerability.
Certain aneuploidies that are commonly found in tumor genomes play a central role in cancer development, and eliminating these aneuploidies compromises malignant growth potential. At the same time, aneuploidy causes collateral therapeutic vulnerabilities that can be targeted to selectively eliminate cells with chromosome dosage imbalances. The development of flexible chromosome engineering methodologies like ReDACT will enable additional experiments to further unravel the consequences of aneuploidy in development and disease.
Most cancers exhibit aneuploidy, but its functional significance in tumor development is controversial. Here, we describe ReDACT (Restoring Disomy in Aneuploid cells using CRISPR Targeting), a set of chromosome engineering tools that allow us to eliminate specific aneuploidies from cancer genomes. Using ReDACT, we created a panel of isogenic cells that have or lack common aneuploidies, and we demonstrate that trisomy of chromosome 1q is required for malignant growth in cancers harboring this alteration. Mechanistically, gaining chromosome 1q increases the expression of MDM4 and suppresses p53 signaling, and we show that TP53 mutations are mutually exclusive with 1q aneuploidy in human cancers. Thus, tumor cells can be dependent on specific aneuploidies, raising the possibility that these “aneuploidy addictions” could be targeted as a therapeutic strategy.
非整倍体,即整个染色体或染色体臂数量的变化,在癌症中很常见,但它们对癌细胞生存的贡献一直难以确定。Girish等人开发了一种染色体工程工具,以协调非整倍体染色体臂的靶向丢失,从而比较带有和不带有选定三体性的同基因癌细胞系。作者发现,特别是染色体1q的三体性对癌细胞有益,并模拟了肿瘤抑制因子TP53信号丢失的表型。具有这种非整倍体的肿瘤对由染色体1q编码的酶激活的化合物敏感,这提示了一种潜在的治疗方法。—Yevgeniya Nusinovich
众所周知,100多年来,人类癌症表现出普遍的非整倍性,即染色体拷贝数变化。例如,约25%的癌症表现出染色体1q臂的增加。然而,尽管非整倍性在多种癌症类型中普遍存在,其在肿瘤发生中的作用仍不明确。我们揭示这些大规模拷贝数改变功能的能力受到无法在癌症中实验性操纵染色体剂量的阻碍。尽管如此,由于非整倍性在恶性肿瘤中常见但在正常组织中罕见,对非整倍体细胞具有选择性毒性的药物可能是有用的抗癌剂。
尽管非整倍体一直难以进行细致分析,先前的研究导致了一种称为“癌基因成瘾”现象的发现。癌基因成瘾的癌症依赖于单个癌基因的表达以持续恶性生长,而该癌基因的丢失或抑制足以诱导癌症消退。由于特定的非整倍体如染色体1q的增加在多种癌症类型中频繁发生,我们假设某些非整倍体本身可能代表类似癌基因的癌症成瘾。为了验证这一假设,我们开发了ReDACT(使用CRISPR靶向恢复非整倍体细胞的二体性),一套染色体工程工具,使我们能够从癌症基因组中消除个体非整倍染色体。使用ReDACT,我们创建并表征了一组具有或缺乏常见癌症非整倍体的同基因细胞。
我们发现,从携带这种改变的癌细胞系中消除染色体1q的三体性几乎完全消除了锚定非依赖性生长和异种移植形成。类似地,从非恶性细胞系中消除1q三体性阻断了RAS介导的转化。在癌细胞系中消除非整倍性后,体外或体内的长期生长导致核型进化,1q二体性细胞最终被恢复1q三体性的细胞所超越。相比之下,从癌细胞中移除其他三体性染色体对恶性生长有不同的影响,表明不同的非整倍体对癌症发展具有不同的表型后果。
临床测序数据的分析表明,染色体1q的增加在肿瘤发生早期出现,并且与肿瘤抑制因子TP53的突变互斥,提示1q三体性可能代表一种不依赖突变的阻断p53信号的机制。与此一致,我们证明了ReDACT介导的染色体1q三体性消除增加了TP53野生型细胞系中p53靶基因的表达。我们将这种p53功能的抑制追溯到MDM4的三倍化,MDM4是编码在染色体1q上的p53抑制剂,并且我们发现删除单个MDM4拷贝损害了1q三体性细胞的生长,而适度过表达MDM4挽救了1q二体性细胞的生长。
最后,我们证明了染色体1q的增加导致UCK2的过表达,UCK2是编码在染色体1q上的核苷酸激酶,也是某些抗癌核苷类似物细胞毒性所必需的。我们确定了几种不同的1q三体性细胞系由于UCK2的上调而对这些化合物表现出增强的敏感性,揭示了1q非整倍性也可能代表一个可处理的癌症脆弱性。
某些在肿瘤基因组中常见的非整倍体在癌症发展中起核心作用,消除这些非整倍体会损害恶性生长潜力。同时,非整倍性引起附带治疗脆弱性,可以靶向选择性消除具有染色体剂量失衡的细胞。灵活染色体工程方法如ReDACT的发展将使得更多实验能够进一步揭示非整倍性在发育和疾病中的后果。
大多数癌症表现出非整倍性,但其在肿瘤发展中的功能意义存在争议。在此,我们描述了ReDACT(使用CRISPR靶向恢复非整倍体细胞的二体性),一套染色体工程工具,使我们能够从癌症基因组中消除特定的非整倍体。使用ReDACT,我们创建了一组具有或缺乏常见非整倍体的同基因细胞,并且我们证明了染色体1q的三体性对于携带这种改变的癌症的恶性生长是必需的。机制上,获得染色体1q增加了MDM4的表达并抑制p53信号,我们显示TP53突变与人类癌症中的1q非整倍性互斥。因此,肿瘤细胞可能依赖于特定的非整倍体,这提出了将这些“非整倍体成瘾”作为治疗策略靶点的可能性。
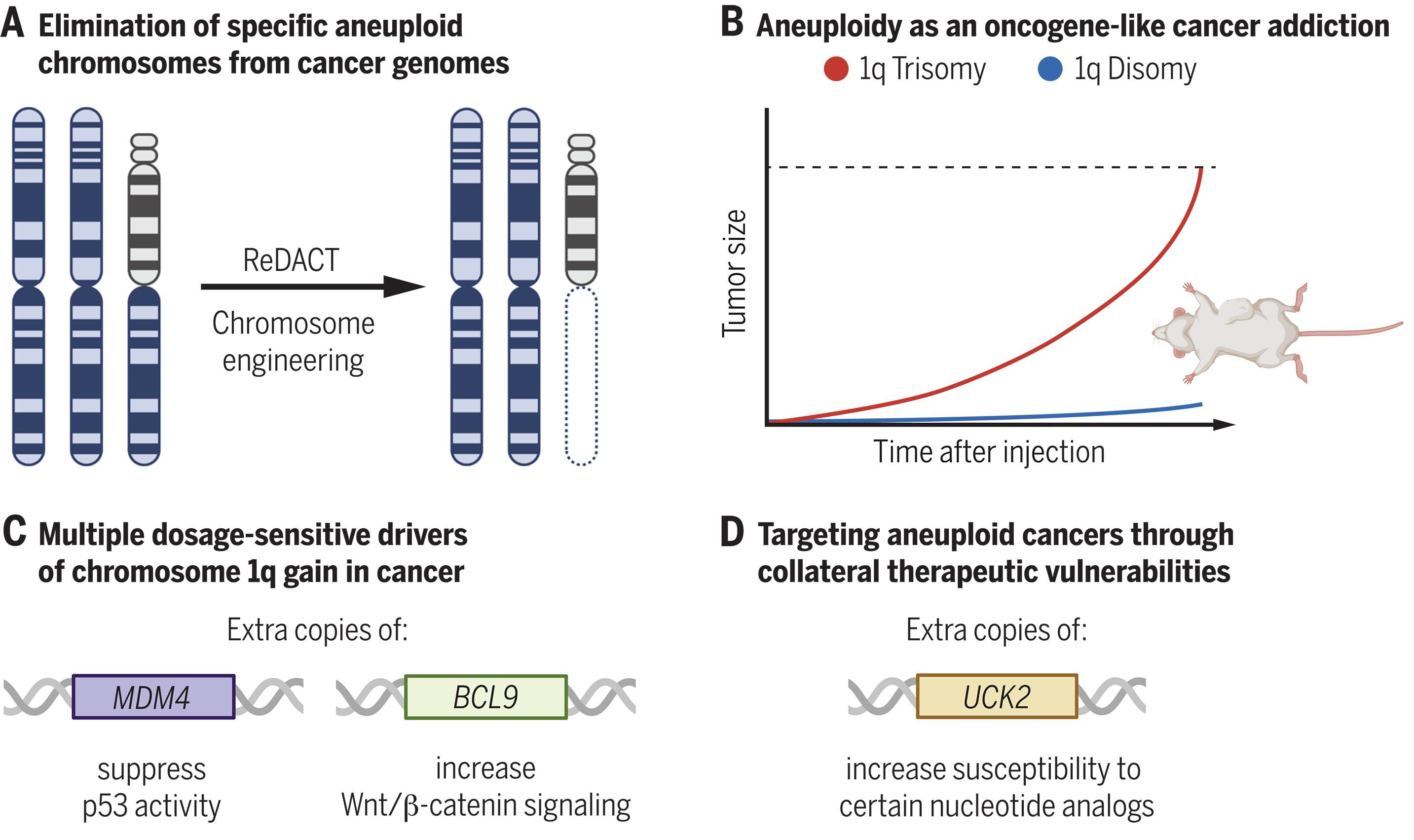
图:染色体工程用于研究非整倍体的影响。(A) ReDACT技术能够靶向删除非整倍体染色体。(B) 额外1号染色体长臂拷贝的缺失会抑制恶性生长。(C) MDM4和BCL9是癌症中1号染色体长臂增益的剂量敏感性驱动因子。(D) 可通过UCK2特异性核苷酸类似物靶向治疗1号染色体长臂增益。