Tumor development is associated with the accumulation of mutations in the genome. Depending on the causes of a given cancer, such as environmental exposures or DNA repair abnormalities, these mutations can form a specific pattern called a mutational signature. Many mutational signatures have already been reported in cancer, but by performing whole-genome sequencing on a particularly large collection of cancer samples, Degasperi et al. not only confirmed previously reported signatures, but also discovered many rarer ones (see the Perspective by Szüts). The authors characterized these signatures, tried to elucidate the underlying biology where possible, and then provided an algorithm for applying these findings to individual patients to help personalize cancer treatments. —YN
Mutational signatures—imprints of DNA damage and repair processes that have been operative during tumorigenesis—provide insights into environmental and endogenous causes of each patient’s cancer. Cancer genome sequencing studies permit exploration of mutational signatures. We investigated a very large number of whole-genome–sequenced cancers of many tumor types, substantially more than in previous efforts, to comprehensively reinforce our understanding of mutational signatures.
We present mutational signature analyses of 12,222 whole-genome–sequenced cancers collected prospectively via the UK National Health Service (NHS) for the 100,000 Genomes Project. We identified single-base substitution (SBS) and double-base substitution (DBS) signatures independently in each organ. Exploiting this unusually large cohort, we developed a method to enhance discrimination of common mutational processes from rare, lower-frequency mutagenic processes. We validated our findings by independently performing analyses with data from two publicly available cohorts: 3001 primary cancers from the International Cancer Genome Consortium (ICGC) and 3417 metastatic cancers from the Hartwig Medical Foundation. We produced a set of reference signatures by comparing and contrasting the independently derived tissue-specific signatures and performing clustering analysis to unite mutational signatures from different tissues that could be due to similar processes. We included additional quality control measures such as dimensionality reduction of mixed signatures and gathered evidence that could help elucidate mechanisms and etiologies such as transcriptional and replication strand bias, associations with somatic drivers, and germline predisposition mutations. We also investigated additional mutation context and examined past clinical and treatment histories when possible, to explore potential etiologies.
Each organ contained a limited number of common SBS signatures (typically between 5 and 10). The number of common signatures was independent of cohort size. By contrast, the number of rare signatures was dependent on sample size, as the likelihood of detecting a rare signature is a function of its population prevalence. The same biological process produced slightly different signatures in diverse tissues, reinforcing that mutational signatures are tissue specific.
Across organs, we clustered all tissue-specific signatures to ascertain mutational processes that were equivalent but occurring in different tissues (i.e., reference signatures). We obtained 82 high-confidence SBS reference signatures and 27 high-confidence DBS reference signatures. We compared these with previously reported mutational signatures, revealing 40 and 18 previously unidentified SBS and DBS signatures, respectively.
Because we are cognizant of increasing complexity in mutational signatures and want to enable general users, we developed an algorithm called Signature Fit Multi-Step (FitMS) that seeks signatures in new samples while taking advantage of our recent findings. In a first step, FitMS detects common, organ-specific signatures; in a second step, it determines whether an additional rare signature is also present.
Mutational signature analysis of 18,640 cancers, the largest cohort of whole-genome–sequenced samples to date, has required methodological advances, permitting knowledge expansion. We have identified many previously unreported signatures and established the concept of common and rare signatures. The FitMS algorithm has been designed to exploit these advances to aid users in accurately identifying mutational processes in new samples.
Whole-genome sequencing (WGS) permits comprehensive cancer genome analyses, revealing mutational signatures, imprints of DNA damage, and repair processes that have arisen in each patient’s cancer. We performed mutational signature analyses on 12,222 whole-genome–sequenced tumor-normal matched pairs from patients recruited via the UK National Health Service (NHS). We contrasted our results with two independent cancer WGS datasets—from the International Cancer Genome Consortium (ICGC) and the Hartwig Medical Foundation (HMF)—involving 18,640 whole-genome–sequenced cancers in total. Our analyses add 40 single and 18 double substitution signatures to the current mutational signature tally. We show for each organ that cancers have a limited number of common signatures and a long tail of rare signatures, and we provide a practical solution for applying this concept of common versus rare signatures to future analyses.
肿瘤的发生发展与基因组中突变的积累有关。根据特定癌症的诱因,例如环境暴露或DNA修复异常,这些突变可以形成一种称为突变特征的特定模式。许多突变特征已在癌症中被报道,但通过对一个数量特别庞大的癌症样本集合进行全基因组测序,Degasperi等人不仅确认了先前报道的特征,还发现了许多更罕见的特征(参见Szüts的观点文章)。作者描述了这些特征,尝试在可能的情况下阐明其背后的生物学机制,随后提供了一种算法,可将这些发现应用于个体患者,以帮助实现癌症治疗的个性化。—YN
突变特征——肿瘤发生过程中起作用的DNA损伤与修复过程的印记——为理解每位患者癌症的环境和内源性诱因提供了见解。癌症基因组测序研究允许对突变特征进行探索。我们研究了大量来自多种肿瘤类型的全基因组测序癌症样本,其数量远超以往研究,旨在全面巩固我们对突变特征的理解。
我们对通过英国国家医疗服务体系为"10万基因组计划"前瞻性收集的12,222例全基因组测序癌症进行了突变特征分析。我们在每个器官中独立识别了单碱基替换(SBS)和双碱基替换(DBS)特征。利用这个异常庞大的队列,我们开发了一种方法来增强对常见突变过程与罕见的、低频致突变过程的区分能力。我们使用来自两个公开可获取队列的数据独立进行分析,验证了我们的发现:来自国际癌症基因组联盟(ICGC)的3001例原发性癌症,以及来自Hartwig医学基金会的3417例转移性癌症。通过比较和对比独立推导的组织特异性特征,并对不同组织中可能由相似过程导致的突变特征进行聚类分析,我们生成了一套参考特征集。我们纳入了额外的质量控制措施,例如混合特征的降维处理,并收集了可能有助于阐明机制和病因的证据,如转录链和复制链偏好、与体细胞驱动基因的关联以及种系易感突变。我们还调查了额外的突变背景,并在可能时查阅了过去的临床和治疗史,以探索潜在的病因。
每个器官都包含数量有限的常见SBS特征(通常在5到10个之间)。常见特征的数量与队列规模无关。相比之下,罕见特征的数量则取决于样本量,因为检测到罕见特征的可能性是其人群流行率的函数。相同的生物学过程在不同的组织中产生了略有不同的特征,这进一步证实了突变特征具有组织特异性。
跨器官层面,我们聚类了所有组织特异性特征,以确定在不同组织中发生但本质相同(即等效)的突变过程(即参考特征)。我们获得了82个高置信度的SBS参考特征和27个高置信度的DBS参考特征。我们将这些与先前报道的突变特征进行比较,分别揭示了40个和18个先前未被识别的SBS和DBS特征。
鉴于我们意识到突变特征日益复杂,并希望赋能普通用户,我们开发了一种名为"特征拟合多步法(FitMS)"的算法。该算法旨在利用我们近期的发现,在新样本中寻找特征。第一步,FitMS检测常见的器官特异性特征;第二步,它判断是否存在额外的罕见特征。
对18,640例癌症(迄今为止最大的全基因组测序样本队列)进行的突变特征分析需要方法学的进步,从而扩展了我们的知识。我们发现了许多先前未报告的特征,并确立了常见特征与罕见特征的概念。FitMS算法的设计正是为了利用这些进展,帮助用户在新样本中准确识别突变过程。
全基因组测序允许进行全面的癌症基因组分析,揭示每位患者癌症中出现的、作为DNA损伤和修复过程印记的突变特征。我们对通过英国国家医疗服务体系招募的患者提供的12,222对肿瘤-正常组织匹配的全基因组测序样本进行了突变特征分析。我们将结果与两个独立的癌症全基因组测序数据集——来自国际癌症基因组联盟(ICGC)和Hartwig医学基金会(HMF)——进行了对比,总共涉及18,640例全基因组测序癌症。我们的分析在当前突变特征总数的基础上增加了40个单碱基替换特征和18个双碱基替换特征。我们展示了对于每个器官,癌症具有有限数量的常见特征和一系列长尾的罕见特征,并提供了一个实用的解决方案,将这种常见与罕见特征的概念应用于未来的分析中。
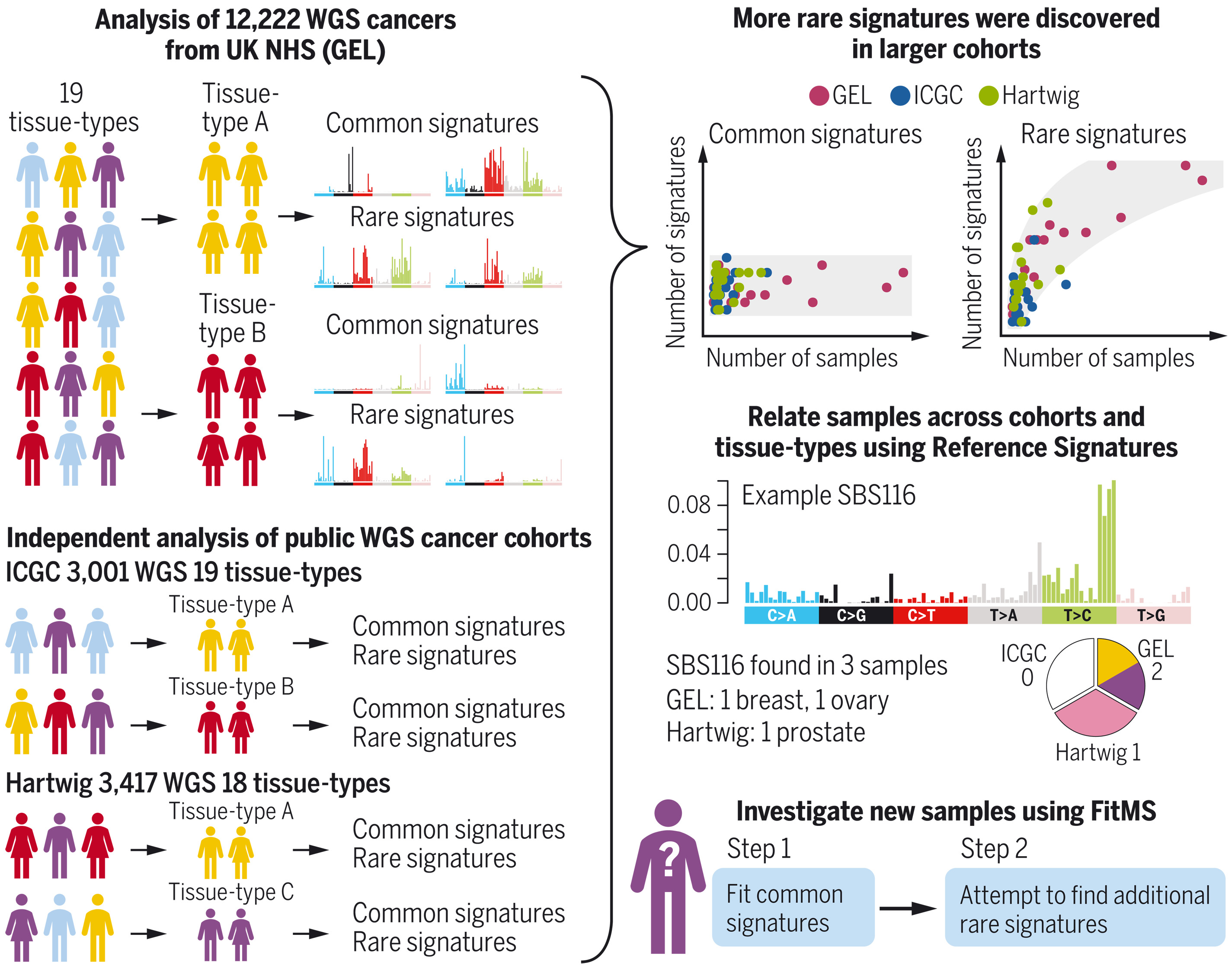
图:常见及罕见突变特征的发现与应用。通过对三个大型全基因组测序癌症队列的分析揭示,各器官常见突变特征数量有限,而罕见特征数量随队列规模扩大而增加。参考特征库支持跨器官和跨队列的比较。此后,一种兼顾常见与罕见特征的新算法FitMS可用于分析新样本。GEL指英国基因组学队列。
Substitution mutational signatures in whole-genome–sequenced cancers in the UK population