Numerous large-scale efforts have been undertaken to catalog and understand the biology of cancer-associated mutations in regions that directly code for proteins. Much of the genome, however, consists of noncoding regions that do not directly encode specific proteins, but instead perform other functions such as regulating protein expression. These genome regions can also play key roles in cancer. Dietlein et al. developed a computational approach to systematically detect cancer-associated mutations in noncoding regions of different cancer types and directly examined the biological function of one such region involved in breast cancer. Using this genome-wide approach, researchers should be able to comprehensively examine the contributions of noncoding regions to cancer development. —YN
A central hallmark of tumor development is that cancer cells acquire somatic mutations in their genomes that are not present in normal tissue. Some mutations are drivers and contribute to the growth of tumor cells, but many others are passengers without apparent effects on tumor biology. Over the past decade, driver mutations have been comprehensively characterized in protein-coding genomic regions by analyzing sequencing data from thousands of tumor-normal pairs. This characterization in protein-coding regions has yielded a wealth of insights into tumor biology, including many genome-inspired drug targets. However, the role of somatic mutations in the other 98% of the cancer genome—the noncoding genome—remains incompletely understood.
Many statistical approaches detect drivers as recurrent mutation events by comparing the number of mutations with and without effects on protein-coding sequences in each gene. These approaches are therefore inapplicable outside of protein-coding regions, where the roles of somatic mutations remain less well understood. The noncoding genome encompasses a diverse spectrum of elements, including regulatory regions of gene expression that differ in their locations and activities between tumor types. To expand our understanding of mutations beyond protein-coding regions, we designed and implemented a genome-wide, sliding-window approach that detects mutation events irrespective of their locations in regulatory elements or effects on protein-coding sequences.
We developed a composite of three methods to detect recurrent mutation events across the whole genomes of 3949 patients with 19 cancer types and 61.2 million somatic mutations. This approach automatically stratified mutation events into different categories on the basis of their position in the genome. In protein-coding regions, we identified an average of 7.5 events per cancer type and recovered well-established driver mutations. In the noncoding genome, 3.7 events per cancer type occurred adjacent to genes exclusively expressed in specific tissue types (ALB in liver, KLK3 in prostate, SFTPB in lung, SLC5A12 in kidney, TG in thyroid tissue, and many others). These tissue-specific events were unlikely to be prototypical drivers because they stemmed from a mutagenic process that was exclusively active around these genes, instead reflecting possible imprints of the expression programs of the tumor cells of origin. Moreover, we found 3.8 noncoding events per cancer type in regulatory regions of expression, many involving cancer-relevant genes (BCL6, FGFR2, RAD51B, SMC6, TERT, XBP1, and many others). In contrast to most events in regulatory regions, breast cancer mutations near XBP1 mainly accumulated in a regulatory region outside of its promoter. We validated their regulatory effects on gene expression by performing CRISPR-interference screening and luciferase reporter assays, illuminating the potential of genome-wide approaches paired with harmonized sequencing cohorts to comprehensively capture mutation patterns in both known and unknown elements of the noncoding genome.
Our study establishes a genome-wide compendium of the diverse mutation patterns that shape the genomes of 19 major cancer types, including events near genes with known roles in tumor biology and some exhibiting experimentally validated effects on gene expression. Our results demonstrate that noncoding mutations are associated with a broad spectrum of different biological processes and that their location in the genome is essential for their accurate interpretation. Broadly, our study provides a blueprint for interpreting whole-genome sequencing data and lays the foundation for future experimental endeavors to implicate noncoding mutations in tumor development, ultimately paving the way for therapies tailored to the noncoding cancer genome.
We established a genome-wide compendium of somatic mutation events in 3949 whole cancer genomes representing 19 tumor types. Protein-coding events captured well-established drivers. Noncoding events near tissue-specific genes, such as ALB in the liver or KLK3 in the prostate, characterized localized passenger mutation patterns and may reflect tumor-cell-of-origin imprinting. Noncoding events in regulatory promoter and enhancer regions frequently involved cancer-relevant genes such as BCL6, FGFR2, RAD51B, SMC6, TERT, and XBP1 and represent possible drivers. Unlike most noncoding regulatory events, XBP1 mutations primarily accumulated outside the gene’s promoter, and we validated their effect on gene expression using CRISPR-interference screening and luciferase reporter assays. Broadly, our study provides a blueprint for capturing mutation events across the entire genome to guide advances in biological discovery, therapies, and diagnostics.
目前已有大量大规模研究工作致力于编录和理解直接编码蛋白质的区域中癌症相关突变的生物学特性。然而,基因组的大部分由非编码区域组成,这些区域不直接编码特定蛋白质,而是执行其他功能,例如调控蛋白质表达。这些基因组区域同样能在癌症中发挥关键作用。Dietlein等人开发了一种计算方法,用于系统性地检测不同癌症类型非编码区域中的癌症相关突变,并直接研究了其中一个涉及乳腺癌的此类区域的生物学功能。利用这种全基因组方法,研究人员应能全面审视非编码区域对癌症发展的贡献。—YN
肿瘤发展的一个核心标志是,癌细胞在其基因组中获得了正常组织不存在的体细胞突变。一些突变是驱动突变,有助于肿瘤细胞的生长,但许多其他突变是乘客突变,对肿瘤生物学没有明显影响。在过去十年中,通过分析数千对肿瘤-正常样本的测序数据,驱动突变在蛋白质编码基因组区域已得到全面表征。这种对蛋白质编码区域的表征为我们理解肿瘤生物学提供了丰富的见解,包括许多受基因组启发的药物靶点。然而,体细胞突变在癌症基因组另外98%的部分——非编码基因组——中的作用仍未得到充分理解。
许多统计方法通过比较每个基因中影响与不影响蛋白质编码序列的突变数量,将复发突变事件检测为驱动突变。因此,这些方法不适用于蛋白质编码区域之外,而在那些区域,体细胞突变的作用仍不甚明了。非编码基因组包含多种多样的元件,包括基因表达的调控区域,这些区域在不同肿瘤类型中的位置和活性各不相同。为了扩展我们对蛋白质编码区域之外突变的理解,我们设计并实施了一种全基因组滑动窗口方法,该方法可检测突变事件,而不论其位于调控元件中的位置或对蛋白质编码序列的影响如何。
我们开发了一种由三种方法组成的复合方法,用于检测代表19种癌症类型的3949名患者的全基因组中的6120万个体细胞突变事件。该方法根据突变事件在基因组中的位置,自动将其划分为不同类别。在蛋白质编码区域,我们平均在每个癌症类型中识别出7.5个事件,并找回了公认的驱动突变。在非编码基因组中,平均每个癌症类型有3.7个事件发生在仅在特定组织类型中表达的基因附近(例如肝脏中的ALB、前列腺中的KLK3、肺中的SFTPB、肾脏中的SLC5A12、甲状腺组织中的TG等)。这些组织特异性事件不太可能是典型的驱动突变,因为它们源于一种仅在这些基因周围活跃的致突变过程,反而可能反映了起源肿瘤细胞表达程序的可能印记。此外,我们在表达调控区域平均每个癌症类型发现了3.8个非编码事件,其中许多涉及癌症相关基因(如BCL6、FGFR2、RAD51B、SMC6、TERT、XBP1等)。与调控区域的大多数事件不同,XBP1附近的乳腺癌突变主要积累在其启动子之外的一个调控区域。我们通过进行CRISPR干扰筛选和荧光素酶报告基因实验验证了它们对基因表达的调控效应,阐明了将全基因组方法与协调一致的测序队列相结合,以全面捕捉非编码基因组中已知和未知元件突变模式的潜力。
我们的研究建立了一个全基因组范围的汇编,涵盖了塑造19种主要癌症类型基因组的多种突变模式,包括肿瘤生物学中已知作用基因附近的事件,以及一些经实验验证对基因表达有影响的事件。我们的结果表明,非编码突变与广泛的多种生物学过程相关联,并且它们在基因组中的位置对其准确解读至关重要。从广义上讲,我们的研究为解读全基因组测序数据提供了蓝图,并为未来将非编码突变与肿瘤发展联系起来的实验工作奠定了基础,最终为针对非编码癌症基因组的疗法铺平了道路。
我们建立了一个全基因组范围的体细胞突变事件汇编,涵盖了代表19种肿瘤类型的3949个完整癌症基因组。蛋白质编码事件捕捉到了公认的驱动突变。组织特异性基因(如肝脏中的ALB或前列腺中的KLK3)附近的非编码事件,表征了局域性乘客突变模式,并可能反映了起源肿瘤细胞的印记。启动子和增强子等调控区域的非编码事件经常涉及癌症相关基因,如BCL6、FGFR2、RAD51B、SMC6、TERT和XBP1,并代表了可能的驱动突变。与大多数非编码调控事件不同,XBP1突变主要积累在该基因的启动子之外,我们利用CRISPR干扰筛选和荧光素酶报告基因实验验证了它们对基因表达的影响。从广义上讲,我们的研究提供了一个捕捉整个基因组突变事件的蓝图,以指导生物学发现、疗法和诊断学的进展。
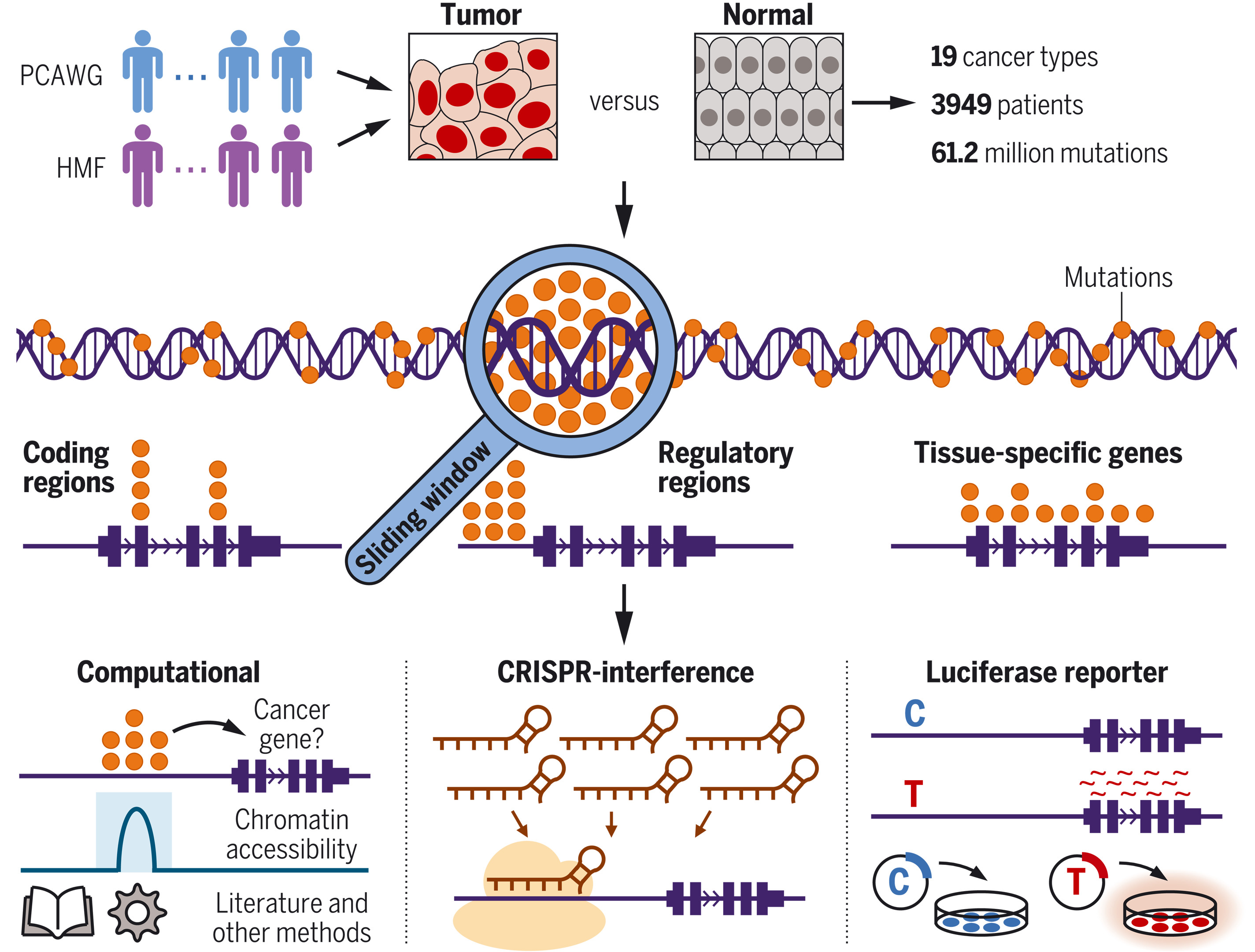
图:人类癌症体细胞突变模式的全基因组目录。我们分析了来自19种癌症类型的3949名患者的6120万个突变(上图)。采用滑动窗口法,我们在整个癌症基因组中检测了突变事件,并按基因组位置对其进行了分类(中图)。为进行系统性后续研究,我们同时采用了计算与实验策略(下图)。PCAWG:全癌种全基因组分析;HMF:哈特维格医学基金会。
Genome-wide analysis of somatic noncoding mutation patterns in cancer