Cancer is a genetic disease, and much cancer research is focused on identifying carcinogenic mutations and determining how they relate to disease progression. Three papers demonstrate how mutations are processed through networks of protein interactions to promote cancer (see the Perspective by Cheng and Jackson). Swaney et al. focus on head and neck cancer and identify cancer-enriched interactions, demonstrating how point mutant–dependent interactions of PIK3CA, a kinase frequently mutated in human cancers, are predictive of drug response. Kim et al. focus on breast cancer and identify two proteins functionally connected to the tumor-suppressor gene BRCA1 and two proteins that regulate PIK3CA. Zheng et al. developed a statistical model that identifies protein networks that are under mutation pressure across different cancer types, including a complex bringing together PIK3CA with actomyosin proteins. These papers provide a resource that will be helpful in interpreting cancer genomic data. —VV
Tumor genome sequencing has revealed that, beyond a few commonly mutated genes, most mutations that affect cancer genomes are rare. To interpret these rare events, a powerful approach has been to organize mutations by their effects on commonly dysregulated cellular systems. Understanding the cancer genome in this way requires surmounting two challenges: (i) How do we comprehensively map cancer cell systems? (ii) How do we identify which systems are under mutational selection?
To address these questions, we used proteomic mass spectrometry and data integration to build a structured map of protein assemblies found in human cancer cells. We then developed a statistical model of mutation, pinpointing which assemblies are under strong mutational selection and in which cancer types. The goal was to interpret the many rare gene mutations that affect tumor genomes by their convergence on higher-order entities.
We amassed a large compendium of cancer protein interactions, combining the screens in breast cancer (Kim et al., this issue) and head-and-neck cancer (Swaney et al., this issue) with multi-omic evidence from 127 previous studies. Lines of evidence were integrated quantitatively to yield a continuous metric of association for each protein pair (integrated association stringency, or IAS). This network of protein associations exhibited clear multiscale and modular structure, revealing 2338 robust assemblies of interacting proteins (hereafter “protein systems”) across different stringencies. Systems were organized hierarchically, with small high-stringency systems (e.g., specific complexes) combining in larger ones (e.g., processes and organelles) as stringency was relaxed.
We next developed a statistical model, HiSig, to identify a parsimonious set of systems that best explains the gene mutation frequencies observed in tumors. HiSig analysis of 13 tumor types yielded a map of 395 mutated protein systems we call NeST (Nested Systems in Tumors, http://ccmi.org/nest/). NeST comprised numerous small complexes, most mutated within specific tumor types, organized within larger systems relevant to most cancers.
Although NeST recapitulated cancer hallmarks, the majority of systems had not been previously described or had not been associated with cancer mutation. Nonetheless, many were recurrently mutated in independent cohorts, supporting their significance. Notable systems included a PIK3CA-actomyosin complex that points to a new mode of phosphatidylinositol 3-kinase regulation, as well as recurrent mutations in collagen complexes that we found to disrupt the extracellular matrix, thereby promoting proliferation. Finally, we identified NeST systems that serve as biomarkers of cancer outcomes, leading to 548 genes for potential use in clinical sequencing panels.
In their classic description of the “Hallmarks of Cancer,” Hanahan and Weinberg predicted that the “complexities of cancer ... will become understandable in terms of a small number of underlying principles.” Around the same time, Alberts provided his seminal perspective of the cell as a collection of “protein assemblies [interacting] in an elaborate network.” By organizing disparate tumor mutations into underlying principles captured by a multiscale map of protein assemblies, this work represents a synthesis of these visions. The strategies developed here may generalize to other diseases that are affected by rare genetic alterations.
A major goal of cancer research is to understand how mutations distributed across diverse genes affect common cellular systems, including multiprotein complexes and assemblies. Two challenges—how to comprehensively map such systems and how to identify which are under mutational selection—have hindered this understanding. Accordingly, we created a comprehensive map of cancer protein systems integrating both new and published multi-omic interaction data at multiple scales of analysis. We then developed a unified statistical model that pinpoints 395 specific systems under mutational selection across 13 cancer types. This map, called NeST (Nested Systems in Tumors), incorporates canonical processes and notable discoveries, including a PIK3CA-actomyosin complex that inhibits phosphatidylinositol 3-kinase signaling and recurrent mutations in collagen complexes that promote tumor proliferation. These systems can be used as clinical biomarkers and implicate a total of 548 genes in cancer evolution and progression. This work shows how disparate tumor mutations converge on protein assemblies at different scales.
癌症是一种基因疾病,许多癌症研究专注于识别致癌突变并确定它们与疾病进展的关系。三篇论文展示了突变如何通过蛋白质相互作用网络被处理从而促进癌症(参见Cheng和Jackson的展望文章)。Swaney等人聚焦于头颈癌,识别出癌症富集的相互作用,展示了PIK3CA(一种在人类癌症中频繁突变的激酶)的点突变依赖性相互作用如何预测药物反应。Kim等人聚焦于乳腺癌,识别出两个与肿瘤抑制基因BRCA1功能相连的蛋白质,以及两个调控PIK3CA的蛋白质。Zheng等人开发了一个统计模型,用于识别在不同癌症类型中承受突变压力的蛋白质网络,其中包括一个将PIK3CA与肌动球蛋白结合起来的复合物。这些论文提供的资源将有助于解读癌症基因组数据。—VV
肿瘤基因组测序显示,除少数常见突变基因外,影响癌症基因组的大多数突变是罕见的。为了解读这些罕见事件,一种强有力的方法是根据突变对通常失调的细胞系统的影响来组织它们。以这种方式理解癌症基因组需要克服两个挑战:(i)我们如何全面绘制癌细胞系统图谱?(ii)我们如何识别哪些系统正处于突变选择之下?
为了解决这些问题,我们利用蛋白质组质谱分析和数据整合,构建了在人类癌细胞中发现的蛋白质组装体的结构化图谱。然后,我们开发了一个突变统计模型,精确定位哪些组装体在强烈的突变选择之下,以及在哪些癌症类型中。其目标是通过将众多罕见的基因突变汇聚到更高层次的实体上,来解读影响肿瘤基因组的这些突变。
我们积累了一个大型的癌症蛋白质相互作用汇编,将乳腺癌(Kim等人,本期)和头颈癌(Swaney等人,本期)的筛选结果与来自127项先前研究的多组学证据相结合。对多线证据进行定量整合,为每个蛋白质对产生一个连续的关联度量指标(整合关联严格性,IAS)。这个蛋白质关联网络展现出清晰的多尺度和模块化结构,揭示了在不同严格性水平上存在的2338个稳健的相互作用蛋白质组装体(下文称“蛋白质系统”)。这些系统按层级组织,随着严格性放宽,小型高严格性系统(例如,特定复合物)会组合成更大的系统(例如,过程和细胞器)。
接下来,我们开发了一个名为HiSig的统计模型,以识别一组最简洁的系统,能够最好地解释在肿瘤中观察到的基因突变频率。对13种肿瘤类型的HiSig分析,产生了一个包含395个突变蛋白质系统的图谱,我们称之为NeST(肿瘤中的嵌套系统,http://ccmi.org/nest/)。NeST包含众多小型复合物,其中大多数在特定肿瘤类型内发生突变,这些复合物被组织在与大多数癌症相关的更大系统中。
尽管NeST重现了癌症的特征,但大多数系统先前未被描述或未与癌症突变相关联。然而,许多系统在独立队列中反复突变,支持了它们的重要性。值得注意的系统包括一个指向磷脂酰肌醇3-激酶调控新模式的PIK3CA-肌动球蛋白复合物,以及我们发现在胶原蛋白复合物中反复发生的突变会破坏细胞外基质,从而促进增殖。最后,我们识别出可作为癌症结果生物标志物的NeST系统,提出了548个潜在可用于临床测序panel的基因。
Hanahan和Weinberg在其经典的“癌症特征”描述中预测,“癌症的复杂性……将能通过少数基本原则来理解。”大约在同一时期,Alberts提出了他开创性的观点,将细胞视为“以精细网络相互作用的蛋白质组装体集合”。通过将分散的肿瘤突变组织到由蛋白质组装体多尺度图谱所捕捉的基本原则中,这项工作代表了这两种愿景的综合。这里开发的策略或可推广到受罕见基因改变影响的其他疾病。
癌症研究的一个主要目标是理解分布在多样基因上的突变如何影响共同的细胞系统,包括多蛋白复合物和组装体。两个挑战——如何全面绘制此类系统图谱以及如何识别哪些系统正处于突变选择之下——阻碍了这种理解。因此,我们创建了一个全面的癌症蛋白质系统图谱,在多个分析尺度上整合了新的和已发表的多组学相互作用数据。然后,我们开发了一个统一的统计模型,精确定位了在13种癌症类型中处于突变选择之下的395个特定系统。这个名为NeST(肿瘤中的嵌套系统)的图谱,包含了经典过程和重要的新发现,包括一个抑制磷脂酰肌醇3-激酶信号传导的PIK3CA-肌动球蛋白复合物,以及促进肿瘤增殖的胶原蛋白复合物中的反复突变。这些系统可用作临床生物标志物,并总共涉及548个基因与癌症进化和进展相关。这项工作展示了不同的肿瘤突变如何在不同尺度上汇聚于蛋白质组装体。
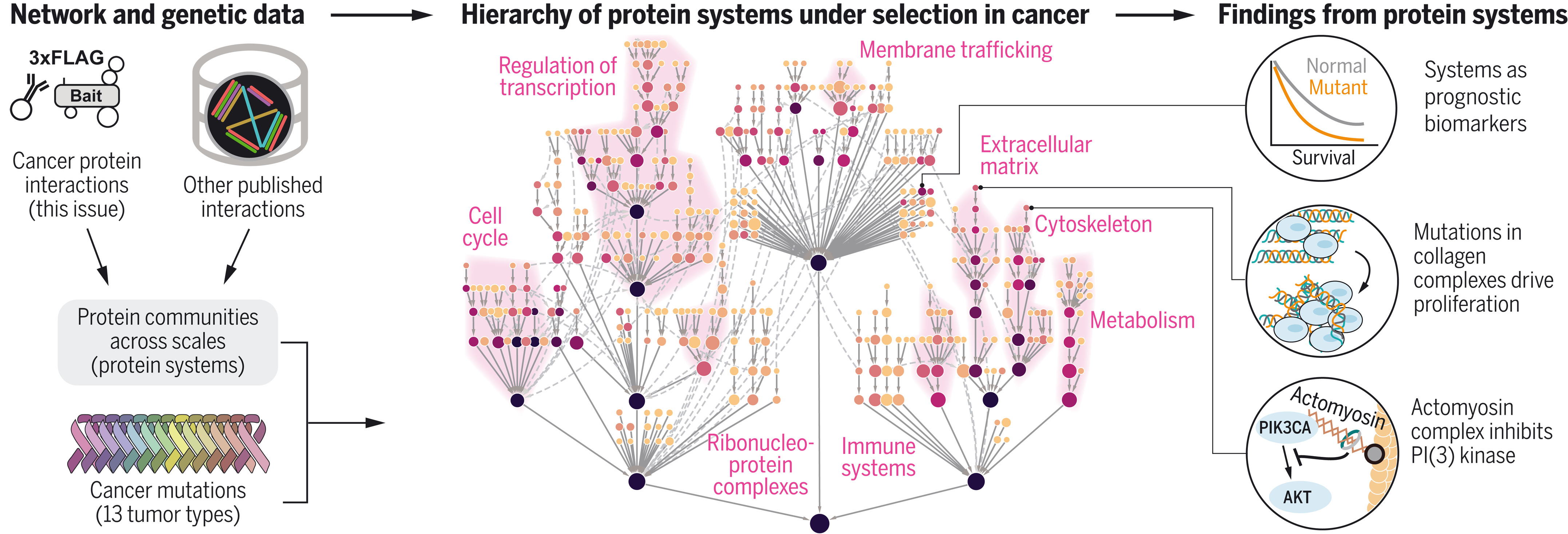
图:癌症蛋白质系统图谱绘制。通过整合蛋白质相互作用数据集,在多个分析尺度上识别蛋白质群落(“系统”)(左图)。针对每个系统进行癌症突变选择测试,以系统整体与其组分蛋白质进行对比,揭示了癌症中选择性作用下蛋白质系统的层级结构(中图)。该层级结构中的发现(右图)已通过临床数据和功能实验得到验证。
Interpretation of cancer mutations using a multiscale map of protein systems