Cancer is a genetic disease, and much cancer research is focused on identifying carcinogenic mutations and determining how they relate to disease progression. Three papers demonstrate how mutations are processed through networks of protein interactions to promote cancer (see the Perspective by Cheng and Jackson). Swaney et al. focus on head and neck cancer and identify cancer-enriched interactions, demonstrating how point mutant–dependent interactions of PIK3CA, a kinase frequently mutated in human cancers, are predictive of drug response. Kim et al. focus on breast cancer and identify two proteins functionally connected to the tumor-suppressor gene BRCA1 and two proteins that regulate PIK3CA. Zheng et al. developed a statistical model that identifies protein networks that are under mutation pressure across different cancer types, including a complex bringing together PIK3CA with actomyosin proteins. These papers provide a resource that will be helpful in interpreting cancer genomic data. —VV
Advances in DNA sequencing technology have enabled the widespread analysis of breast tumor genomes, creating a catalog of genetic mutations that may initiate or drive tumor progression. In addition to common mutations in well-known cancer genes, such as TP53 and PIK3CA, breast cancers harbor a variety of rare mutations with low prevalence across the patient population. Despite this heterogeneity, the majority of breast cancer patients are treated using broad chemotherapy or hormone therapies, which vary widely in effectiveness across patients. Therefore, there is an urgent need to develop targeted therapies matched to the specific molecular alterations in each patient’s tumor, with the goal of improving efficacy, reducing toxicity, and avoiding unnecessary treatment.
A key question is how these rare alterations elicit pathologic consequences, control patient outcomes, and, ultimately, translate into personalized therapies. An answer lies in understanding how individual gene mutations converge on multigene functional modules, including the signaling pathways that orchestrate cell proliferation, apoptosis, and DNA repair. To broadly enable a pathway-based understanding of cancer, we must first generate comprehensive maps of cancer molecular networks in relevant malignant and premalignant cellular contexts.
To this end, we used affinity purification combined with mass spectrometry (AP-MS) to catalog protein-protein interactions (PPIs) for 40 proteins significantly altered in breast cancer, including multidimensional measurements across mutant and normal protein isoforms and across cancerous and noncancerous cellular contexts. Approximately 79% of the PPIs that we identified have not been previously reported, and 81% are not shared across cell lines, which illustrates a substantial rewiring of PPIs driven by different cellular contexts. Notably, interacting proteins specific to two breast cancer cell lines (MCF7 and MDA-MB-231) are more frequently mutated in breast tumors than interacting proteins recovered in nontumorigenic MCF10A cells, which implies that proteins interacting with known cancer drivers may also contribute to the onset of cancer.
AP-MS analysis of PIK3CA identified previously unidentified interacting proteins (BPIFA1 and SCGB2A1) that act as potent negative regulators of the PI3K-AKT pathway in multiple breast cancer cell contexts, providing new mechanistic and therapeutic insights into the regulation of this key signaling pathway. Furthermore, UBE2N emerged as a functionally relevant interactor of BRCA1, and we show that its expression could serve as a potential biomarker of response to poly(ADP-ribose) polymerase (PARP) inhibitors and other DNA repair targeted therapies. We also found that the protein phosphatase 1 (PP1) regulatory subunit spinophilin interacts with and regulates dephosphorylation of BRCA1 and other DNA repair proteins to promote DNA double-strand break repair.
Our study demonstrates that systematic PPI maps provide a useful resource in contextualizing uncharacterized mutations within signaling pathways and protein complexes. Such maps effectively identify previously unidentified cancer susceptibility genes and druggable vulnerabilities in not only breast cancer but head and neck cancer as well (Swaney et al., this issue). These efforts are informing hierarchical maps of protein complexes and systems in both healthy and diseased cells (Zheng et al., this issue), which can be used to stratify patients for known anticancer therapies and drive the discovery of therapeutic targets for cancer as well as a variety of other diseases.
Cancers have been associated with a diverse array of genomic alterations. To help mechanistically understand such alterations in breast-invasive carcinoma, we applied affinity purification–mass spectrometry to delineate comprehensive biophysical interaction networks for 40 frequently altered breast cancer (BC) proteins, with and without relevant mutations, across three human breast cell lines. These networks identify cancer-specific protein-protein interactions (PPIs), interconnected and enriched for common and rare cancer mutations, that are substantially rewired by the introduction of key BC mutations. Our analysis identified BPIFA1 and SCGB2A1 as PIK3CA-interacting proteins, which repress PI3K-AKT signaling, and uncovered USP28 and UBE2N as functionally relevant interactors of BRCA1. We also show that the protein phosphatase 1 regulatory subunit spinophilin interacts with and regulates dephosphorylation of BRCA1 to promote DNA double-strand break repair. Thus, PPI landscapes provide a powerful framework for mechanistically interpreting disease genomic data and can identify valuable therapeutic targets.
癌症是一种基因疾病,大量癌症研究专注于识别致癌突变并确定其与疾病进展的关系。三篇论文展示了突变如何通过蛋白质相互作用网络进行处理以促进癌症(参见Cheng和Jackson的观点文章)。Swaney等人聚焦头颈癌,识别出癌症富集的相互作用,展示了PIK3CA(一种在人类癌症中频繁突变的激酶)的点突变依赖性相互作用如何预测药物反应。Kim等人聚焦乳腺癌,识别出两个与抑癌基因BRCA1功能连接的蛋白质,以及两个调控PIK3CA的蛋白质。Zheng等人开发了一个统计模型,识别出在不同癌症类型中承受突变压力的蛋白质网络,其中包括一个将PIK3CA与肌动球蛋白结合起来的复合物。这些论文提供了一个有助于解读癌症基因组数据的资源库。—VV
DNA测序技术的进步使得对乳腺肿瘤基因组进行广泛分析成为可能,从而创建了一个可能引发或驱动肿瘤进展的基因突变目录。除了TP53和PIK3CA等著名癌症基因中的常见突变外,乳腺癌还含有多种在患者群体中流行率较低的罕见突变。尽管存在这种异质性,大多数乳腺癌患者仍使用广泛的化疗或激素疗法进行治疗,这些疗法在不同患者中的疗效差异很大。因此,迫切需要开发针对每位患者肿瘤中特定分子改变的特异性疗法,旨在提高疗效、降低毒性并避免不必要的治疗。
一个关键问题是这些罕见的改变如何引发病理后果、控制患者结局,并最终转化为个体化疗法。答案在于理解个体基因突变如何汇聚于多基因功能模块,包括协调细胞增殖、凋亡和DNA修复的信号通路。为了广泛实现基于通路的癌症理解,我们必须首先在相关的恶性和癌前细胞环境中生成全面的癌症分子网络图谱。
为此,我们采用亲和纯化结合质谱法(AP-MS)来编录40种在乳腺癌中显著改变的蛋白质的蛋白质-蛋白质相互作用(PPI),包括跨突变体和正常蛋白质异构体以及跨癌性和非癌性细胞环境的多维度测量。我们鉴定出的PPI中约有79%此前未被报道,81%在不同细胞系间不共享,这说明了由不同细胞环境驱动的PPI发生了重大的重排。值得注意的是,在两种乳腺癌细胞系(MCF7和MDA-MB-231)中特有的相互作用蛋白,比在非致瘤性MCF10A细胞中回收的相互作用蛋白,在乳腺肿瘤中更频繁地发生突变,这意味着与已知癌症驱动蛋白相互作用的蛋白也可能促进癌症的发生。
对PIK3CA的AP-MS分析发现了先前未知的相互作用蛋白(BPIFA1和SCGB2A1),它们在多种乳腺癌细胞环境中充当PI3K-AKT通路有效的负调控因子,为这一关键信号通路的调控提供了新的机制和治疗见解。此外,UBE2N作为BRCA1的功能相关相互作用蛋白出现,我们表明其表达可作为对聚(ADP-核糖)聚合酶(PARP)抑制剂和其他DNA修复靶向疗法反应的潜在生物标志物。我们还发现,蛋白磷酸酶1(PP1)的调节亚基spionophilin与BRCA1及其他DNA修复蛋白相互作用并调节其去磷酸化,从而促进DNA双链断裂修复。
我们的研究表明,系统的PPI图谱为在信号通路和蛋白质复合物中定位未表征的突变提供了一个有用的资源。此类图谱不仅能有效识别先前未知的癌症易感基因和可药物靶点弱点,不仅针对乳腺癌,也针对头颈癌(本期Swaney等人文章)。这些工作正在构建健康和病变细胞中蛋白质复合物和系统的层级图谱(本期Zheng等人文章),这些图谱可用于为已知的抗癌疗法对患者进行分层,并推动癌症以及多种其他疾病治疗靶点的发现。
癌症与多种基因组改变相关。为了从机制上理解乳腺浸润性癌中的此类改变,我们应用亲和纯化-质谱联用技术,在三种人类乳腺细胞系中,为40种频繁改变的乳腺癌(BC)蛋白(包含或不包含相关突变)描绘了全面的生物物理相互作用网络。这些网络识别出癌症特异性的蛋白质-蛋白质相互作用(PPI),它们相互连接并富含常见和罕见的癌症突变,并且会因引入关键的BC突变而发生显著的重排。我们的分析确定BPIFA1和SCGB2A1为PIK3CA的相互作用蛋白,它们抑制PIK3-AKT信号传导;并发现USP28和UBE2N是BRCA1的功能相关相互作用蛋白。我们还表明,蛋白磷酸酶1调节亚基spionophilin与BRCA1相互作用并调节其去磷酸化以促进DNA双链断裂修复。因此,PPI图谱为从机制上解读疾病基因组数据提供了一个强大的框架,并能识别有价值的治疗靶点。
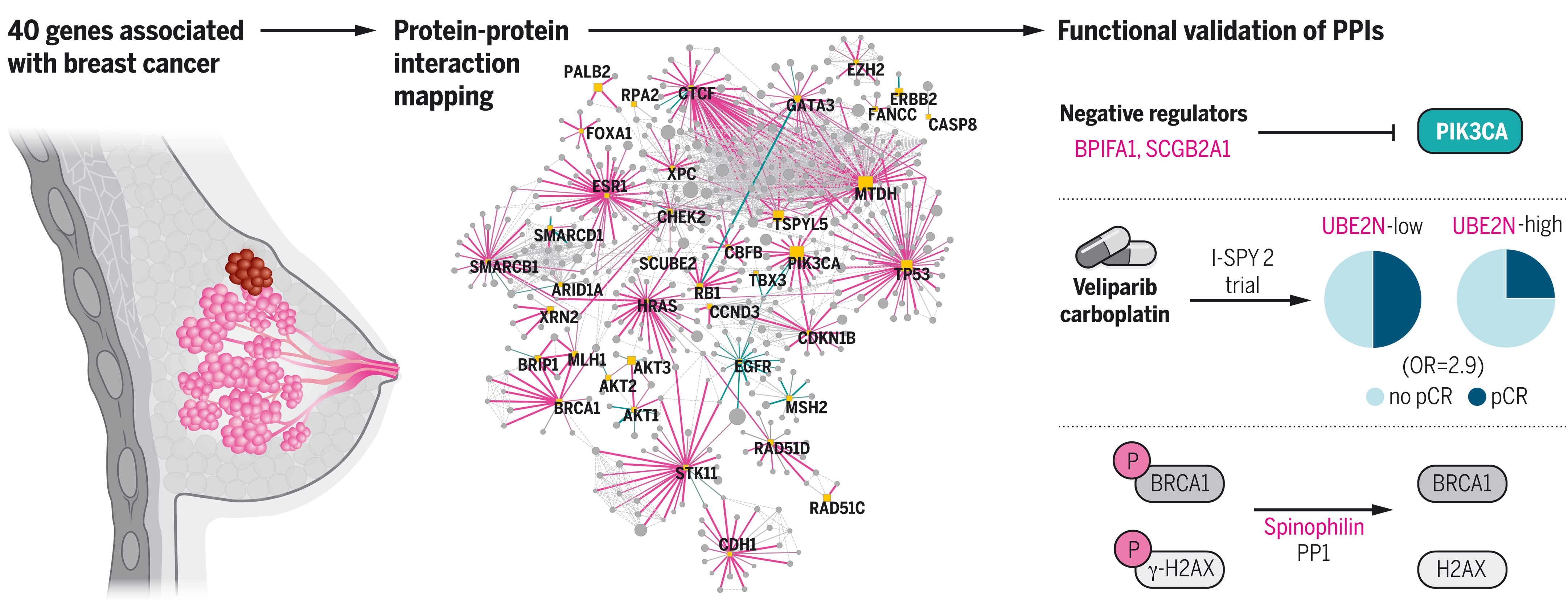
图:胰腺癌中的隐秘抗原。非编码基因的异常翻译会产生非常规(隐秘)肽段,这些肽段能够在胰腺癌中被 HLA-I 处理并呈递。隐秘肽段具有很强的免疫原性,相应的 TCR 能够在体外和体内识别并杀死患者来源的胰腺癌细胞团。nuORF,新未注释的开放阅读框;dORF,下游开放阅读框;Ribo-seq,核糖体测序;CTLs,细胞毒性 T 淋巴细胞;IFN-γ,干扰素-γ;TNFα,肿瘤坏死因子-α。