Many human tumors display scrambled genomes that arise from two distinct mutational processes. The first, the chromosome breakage-fusion-bridge (BFB) cycle, produces gene amplification and genomic instability. The second, chromothripsis, generates massive, clustered genomic rearrangements in one or a few chromosomes. Umbreit et al. hypothesized that these two processes are mechanistically related and tested this idea by recreating essential steps of the BFB cycle in cultured cells (see the Perspective by Paiano and Nussenzweig). They found that chromothripsis arises from a cascade of events that begins with aberrant chromosome bridge formation during mitosis, followed by chromosome fragmentation, DNA damage, chromosome missegregation, and the formation of micronuclei. They propose a model that explains how a single cell division error (chromosome bridge formation) can generate many hallmark features of cancer genomes.
The chromosome breakage-fusion-bridge (BFB) cycle is a catastrophic mutational process, common during tumorigenesis, that results in gene amplification and drives rapid genome evolution. Major mechanisms underlying the BFB cycle are not understood, including its key feature of how chromosome bridges are broken. Furthermore, the simple pattern of DNA sequence rearrangement predicted by the canonical BFB model is not commonly observed in cancer genomes. Instead, the DNA sequence signature of BFB cycles is often accompanied by other genomic rearrangements, including chromothripsis, another catastrophic mutational pattern.
We recreated essential steps of the BFB cycle in a defined system, enabling mechanistic studies and determination of the immediate and long-term genomic consequences of bridge formation. To identify the immediate outcomes of bridge breakage, we used live-cell imaging coupled with single-cell whole-genome sequencing (Look-Seq). Complex mutational mechanisms, some of which occurred over two generations, could be deconvolved by the comparison of haplotype copy number and structural variants in daughter or granddaughter cells. We then determined the long-term consequences of bridge breakage with genomic analysis of populations derived from single cells after breakage of a bridge formed from an experimentally induced dicentric fusion of chromosome 4.
We showed that chromosome bridge breakage requires actomyosin-dependent mechanical force. Bridge formation and breakage is then coupled to a cascade of additional mutational events. For the initial step, we determined that direct mechanical bridge breakage can generate simple breaks and local DNA fragmentation, providing one explanation for a rearrangement pattern frequently observed in cancer genomes termed “local jumps.” Concomitantly, there is defective DNA replication of bridge DNA, which our data suggest can generate complex rearrangements. Some of these rearrangements exhibit a distinct sequence signature of tandem arrays of many short (~200 base pairs) insertions that we term “Tandem Short Template (TST) jumps.” We validated the presence of TST jumps in a human cancer by use of single-molecule long-read DNA sequencing. Next, a second wave of DNA damage and increased chromothripsis occurs during the mitosis after bridge formation, when chromosomes from broken bridges undergo an unexpected burst of aberrant DNA replication. Last, these damaged bridge chromosomes missegregate with high frequency and form micronuclei in the following cell cycle, which can generate additional cycles of bridging, micronucleation, and chromothripsis. Genome sequence analysis of clonal populations established that the breakage of chromosome bridges initiates iterative cycles of complex karyotype evolution. We observed an analogous series of events after the formation of micronuclei, suggesting a unifying model for how cancer-associated defects in nuclear architecture (“nuclear atypia”) promote genome instability.
We identified a cascade of events that explains how a single cell division error—chromosome bridge formation—can rapidly generate many hallmark features of cancer genomes, including ongoing genome evolution with subclonal heterogeneity. These results motivate a substantial revision of the chromosome BFB model, establishing that episodes of chromothripsis will be inherently interwoven with BFB cycles. These mutational events are common in cancer but likely also occur during development and across organismal evolution.
The chromosome breakage-fusion-bridge (BFB) cycle is a mutational process that produces gene amplification and genome instability. Signatures of BFB cycles can be observed in cancer genomes alongside chromothripsis, another catastrophic mutational phenomenon. We explain this association by elucidating a mutational cascade that is triggered by a single cell division error—chromosome bridge formation—that rapidly increases genomic complexity. We show that actomyosin forces are required for initial bridge breakage. Chromothripsis accumulates, beginning with aberrant interphase replication of bridge DNA. A subsequent burst of DNA replication in the next mitosis generates extensive DNA damage. During this second cell division, broken bridge chromosomes frequently missegregate and form micronuclei, promoting additional chromothripsis. We propose that iterations of this mutational cascade generate the continuing evolution and subclonal heterogeneity characteristic of many human cancers.
许多人类肿瘤表现出混乱的基因组,这些基因组源于两种不同的突变过程。第一种是染色体断裂-融合-桥(BFB)循环,导致基因扩增和基因组不稳定。第二种是染色体碎裂,在一个或少数几个染色体上产生大量聚集的基因组重排。Umbreit等人假设这两个过程在机制上相关,并通过在培养细胞中重现BFB循环的关键步骤来验证这一想法(参见Paiano和Nussenzweig的观点)。他们发现染色体碎裂源于一系列事件,始于有丝分裂过程中异常染色体桥的形成,随后是染色体片段化、DNA损伤、染色体错误分离和微核的形成。他们提出了一个模型,解释了一个细胞分裂错误(染色体桥形成)如何能够产生癌症基因组的许多标志性特征。
染色体断裂-融合-桥(BFB)循环是一种灾难性的突变过程,在肿瘤发生过程中常见,导致基因扩增并驱动快速的基因组进化。BFB循环的基本机制尚不清楚,包括其关键特征——染色体桥是如何断裂的。此外,经典BFB模型预测的简单DNA序列重排模式在癌症基因组中并不常见。相反,BFB循环的DNA序列特征通常伴随其他基因组重排,包括另一种灾难性突变模式——染色体碎裂。
我们在一个确定的系统中重建了BFB循环的关键步骤,从而能够进行机制研究,并确定桥形成的即时和长期基因组后果。为了确定桥断裂的直接结果,我们使用了活细胞成像结合单细胞全基因组测序(Look-Seq)。复杂的突变机制,其中一些发生在两代之间,可以通过比较子细胞或孙细胞中的单倍型拷贝数和结构变异来解析。然后,我们通过对由实验诱导的4号染色体双着丝粒融合形成的桥断裂后的单细胞衍生群体进行基因组分析,确定了桥断裂的长期后果。
我们证明了染色体桥断裂需要依赖于肌动球蛋白的机械力。桥的形成和断裂随后与一系列额外的突变事件相关联。对于初始步骤,我们确定直接的机械桥断裂可以产生简单的断裂和局部DNA片段化,这为癌症基因组中经常观察到的一种重排模式——“局部跳跃”提供了一种解释。同时,桥DNA的DNA复制存在缺陷,我们的数据表明这可以产生复杂的重排。其中一些重排表现出许多短(约200个碱基对)插入的串联阵列的独特序列特征,我们称之为“串联短模板(TST)跳跃”。我们通过使用单分子长读长DNA测序验证了人类癌症中TST跳跃的存在。接下来,在桥形成后的有丝分裂过程中,当来自断裂桥的染色体经历意外的异常DNA复制爆发时,发生了第二波DNA损伤和增加的染色体碎裂。最后,这些受损的桥染色体以高频率错误分离,并在随后的细胞周期中形成微核,这可以产生额外的桥接、微核形成和染色体碎裂循环。对克隆群体的基因组序列分析确定,染色体桥的断裂启动了复杂核型进化的迭代循环。我们在微核形成后观察到类似的一系列事件,提出了一个统一的模型,解释癌症相关的核结构缺陷(“核异型”)如何促进基因组不稳定。
我们确定了一系列事件,解释了一个细胞分裂错误——染色体桥形成——如何能够快速产生癌症基因组的许多标志性特征,包括具有亚克隆异质性的持续基因组进化。这些结果促使对染色体BFB模型进行重大修订,确定染色体碎裂事件将与BFB循环内在地交织在一起。这些突变事件在癌症中很常见,但也可能在发育过程中和整个生物体进化中发生。
染色体断裂-融合-桥(BFB)循环是一种突变过程,导致基因扩增和基因组不稳定。BFB循环的特征可以在癌症基因组中与另一种灾难性突变现象——染色体碎裂一起观察到。我们通过阐明由一个细胞分裂错误(染色体桥形成)触发的突变级联反应来解释这种关联,该错误迅速增加了基因组的复杂性。我们表明肌动球蛋白力是初始桥断裂所必需的。染色体碎裂累积,始于桥DNA的异常间期复制。随后在下一个有丝分裂中的DNA复制爆发产生广泛的DNA损伤。在第二次细胞分裂期间,断裂的桥染色体经常错误分离并形成微核,促进额外的染色体碎裂。我们提出,这种突变级联的迭代产生了许多人类癌症的特征——持续进化和亚克隆异质性。
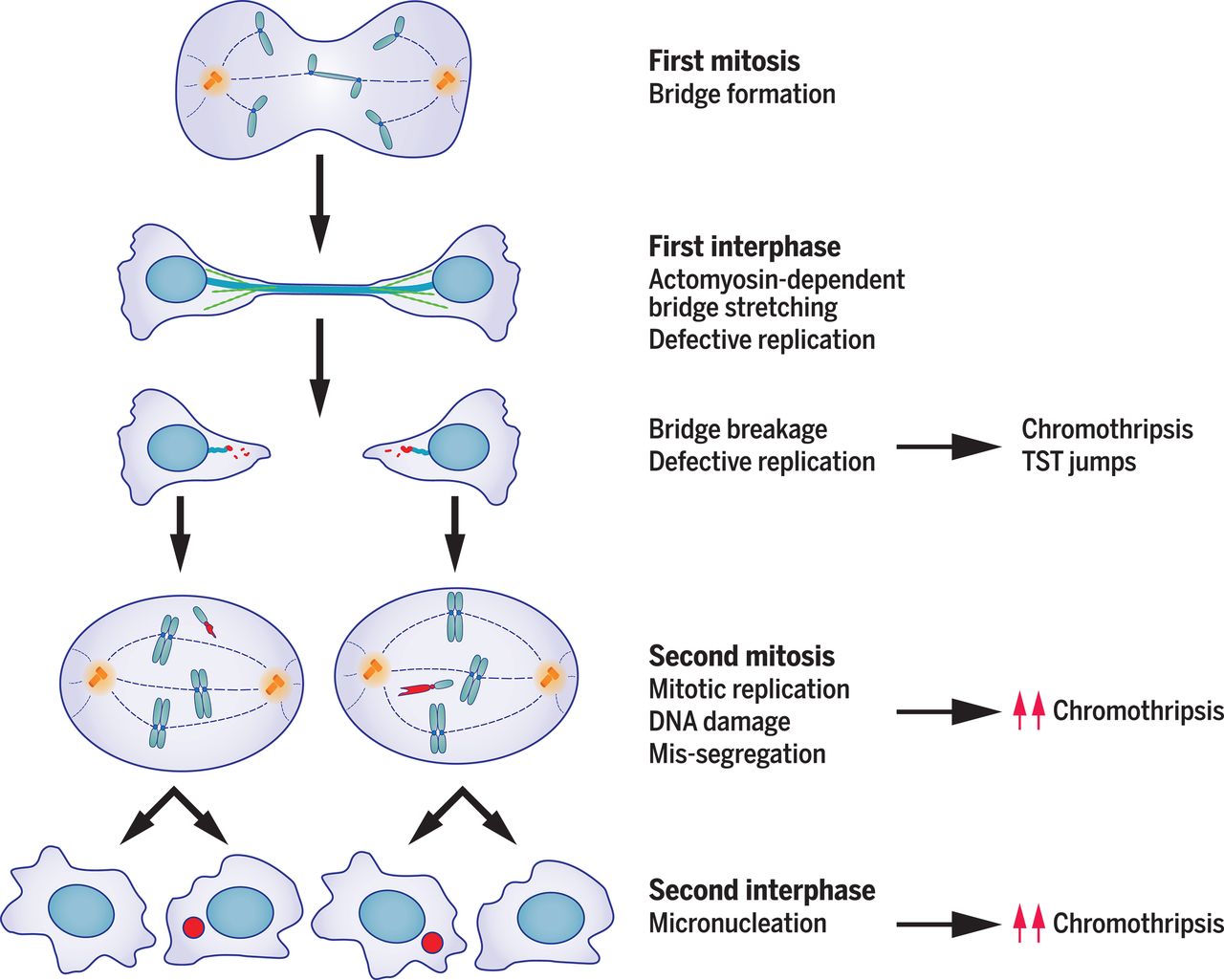
图:一次细胞分裂错误引发的突变风暴,造就了癌症基因组的复杂性。分裂间期的肌动蛋白细胞骨架(绿色纤维)拉伸并断裂染色体桥,促进局部染色体碎裂(受损DNA以红色标记)。从分裂间期到随后有丝分裂过程中出现的DNA复制缺陷,会引发额外DNA损伤与染色体碎裂,有时会留下特异的突变特征(TST跳跃)。断裂的染色体常发生错误分离并形成微核,导致更多染色体碎裂。
Mechanisms generating cancer genome complexity from a single cell division error