Cancer cells that give rise to solid tumors reside in a specialized microenvironment containing growth factors that support tumor growth and progression. The mechanisms by which tumor cells within this so-called “niche” attract and sustain these growth factors are poorly understood. Studying a mouse model of squamous cell carcinoma, Taniguchi et al. identified a signaling loop initiated by certain tumor cells within the niche that become more invasive on exposure to transforming growth factor β (TGF-β). They show that the tumor cells release the cytokine interleukin-33, which induces the differentiation of nearby myeloid cells into macrophages. These macrophages in turn release TGF-β, which feeds back to the tumor cells, promoting their malignant progression.
A small subset of tumor cells with long-term tumorigenic capacity, known as tumor-initiating cells (TICs), play a pivotal role in cancer development and therapy resistance. However, the development of effective TIC-targeted therapies is moving at a restricted pace due to the lack of identification of TIC vulnerabilities. Just as normal stem cells are regulated by external cues derived from specialized microenvironments, or stem cell niches, the stem-like state of TICs and the malignant phenotypes of their progeny are controlled by various factors emanating from the TIC-associated tumor microenvironment, the so-called TIC niche. Therefore, a mechanistic understanding of the cross-talk between TICs and the niche could accelerate the development of durable cancer therapeutics. Although the TIC niche is thought to evolve through reciprocal interactions with TICs, the mechanism by which the TIC–niche interaction emerges in the course of tumor development is poorly understood. Solid tumors are known to recruit immune cells in the stroma and create favorable conditions for their growth and survival. However, not much is known about how TICs regulate the localization and function of TIC-supportive immune cells in their spatial proximity.
Using a mouse model of squamous cell carcinoma (SCC), we previously showed that transforming growth factor β (TGF-β) induces a subset of drug-resistant TICs that give rise to invasive, poorly differentiated progeny. We observed that these TGF-β–responding tumor cells are spatially associated with localized TGF-β expression in the adjacent stroma. Therefore, the mechanisms that lead to “TGF-β–rich” tumor microenvironments may underlie the development of TIC–niche interactions and potentially be exploited as a new target for destabilizing TICs. Because normal stem cells coordinate their niches by sending short-distance signals, we hypothesized that TICs might send a specific signaling molecule to the adjacent stroma to induce a TIC-supporting niche.
Focusing on the cytokine milieu and immune cells in the proximity of TGF-β–responding TICs, here, we address how TICs generate a spatially distinct niche microenvironment that is required for invasive progression and drug resistance of SCC. In a search for potential paracrine regulators of the neighboring tumor microenvironment, we identified interleukin-33 (IL-33) as the most highly up-regulated cytokine in TGF-β–responding TICs. Whereas IL-33 is stored in the nucleus under normal conditions, we found that it is released into the extracellular space in response to the NRF2-mediated antioxidant response, a hallmark of TGF-β–responding TICs. This TIC-derived IL-33 was required for invasive progression and drug resistance of SCC. Mechanistically, IL-33 induces the accumulation of a subset of tumor-associated macrophages expressing the IL-33 receptor ST2 and the high-affinity IgE receptor (FcεRIα) in close proximity to TICs (i.e., within a 50-μm radius). These previously unappreciated FcεRIα+ macrophages were differentiated and alternatively activated from bone marrow–derived cells and created a TGF-β–rich niche microenvironment through the IL-33–ST2–NF-κB pathway, inducing paracrine TGF-β signaling to TICs and further upregulating IL-33 expression. The abrogation of the pathway or the depletion of FcεRIα+ macrophages reduced the number of TGF-β–responding TICs, the rate of invasive tumor progression, and chemotherapy resistance.
Therapy-resistant TICs are considered to be major culprits in cancer treatment failure. Studying a mouse model, we unveil the cellular and molecular basis for the formation of a TIC niche that promotes malignant progression and drug resistance of SCC. The discovery of the IL-33–TGF-β niche signaling loop between TICs and FcεRIα+ macrophages provides mechanistic insights into self-reinforcing TIC–niche interactions, which could be a potential target for destabilizing TICs to improve cancer treatment efficacy.
Targeting the cross-talk between tumor-initiating cells (TICs) and the niche microenvironment is an attractive avenue for cancer therapy. We show here, using a mouse model of squamous cell carcinoma, that TICs play a crucial role in creating a niche microenvironment that is required for tumor progression and drug resistance. Antioxidant activity in TICs, mediated by the transcription factor NRF2, facilitates the release of a nuclear cytokine, interleukin-33 (IL-33). This cytokine promotes differentiation of macrophages that express the high-affinity immunoglobulin E receptor FcεRIα and are in close proximity to TICs. In turn, these IL-33–responding FcεRIα+ macrophages send paracrine transforming growth factor β (TGF-β) signals to TICs, inducing invasive and drug-resistant properties and further upregulating IL-33 expression. This TIC-driven, IL-33–TGF-β feedforward loop could potentially be exploited for cancer treatment.
产生实体瘤的癌细胞存在于一个特殊的微环境中,其中含有支持肿瘤生长和进展的生长因子。肿瘤细胞在这个所谓的“微环境”中如何吸引并维持这些生长因子的机制尚不清楚。通过研究鳞状细胞癌小鼠模型,Taniguchi等人发现了一个由微环境中某些肿瘤细胞启动的信号循环,这些肿瘤细胞在暴露于转化生长因子β(TGF-β)后变得更具侵袭性。他们表明,肿瘤细胞释放细胞因子白细胞介素-33,诱导附近的髓样细胞分化为巨噬细胞。这些巨噬细胞反过来释放TGF-β,反馈给肿瘤细胞,促进其恶性进展。
一小部分具有长期致瘤能力的肿瘤细胞,称为肿瘤起始细胞(TICs),在癌症发展和治疗耐药中起关键作用。然而,由于尚未识别TICs的弱点,有效的靶向TICs疗法的开发进展缓慢。正如正常干细胞受到来自特殊微环境(即干细胞微环境)的外部信号调控一样,TICs的干细胞样状态及其后代的恶性表型受到TICs相关肿瘤微环境(即TICs微环境)产生的多种因子控制。因此,理解TICs与微环境之间相互作用的机制可能加速持久性癌症疗法的开发。尽管TICs微环境被认为通过与TICs的相互作用而演变,但TICs-微环境相互作用在肿瘤发展过程中出现的机制尚不清楚。已知实体瘤会招募基质中的免疫细胞,为其生长和存活创造有利条件。然而,关于TICs如何调节其空间邻近的支持性免疫细胞的定位和功能,目前知之甚少。
使用鳞状细胞癌(SCC)小鼠模型,我们先前证明转化生长因子β(TGF-β)诱导了一部分耐药TICs,这些TICs产生侵袭性、低分化的后代。我们观察到这些对TGF-β反应的肿瘤细胞在空间上与邻近基质中局部TGF-β表达相关。因此,导致“富含TGF-β”肿瘤微环境的机制可能是TICs-微环境相互作用发展的基础,并可能被用作破坏TICs稳定性的新靶点。由于正常干细胞通过发送短距离信号来协调其微环境,我们假设TICs可能向邻近基质发送特定的信号分子,以诱导支持TICs的微环境。
着眼于对TGF-β反应的TICs附近的细胞因子环境和免疫细胞,我们在此探讨TICs如何产生空间上独特的微环境,该微环境是SCC侵袭性进展和耐药所必需的。在寻找邻近肿瘤微环境的潜在旁分泌调节因子时,我们确定白细胞介素-33(IL-33)是在对TGF-β反应的TICs中上调最显著的细胞因子。虽然在正常条件下IL-33储存在细胞核中,但我们发现它响应于NRF2介导的抗氧化反应(对TGF-β反应的TICs的标志)而被释放到细胞外空间。这种TICs来源的IL-33是SCC侵袭性进展和耐药所必需的。机制上,IL-33诱导表达IL-33受体ST2和高亲和力IgE受体(FcεRIα)的肿瘤相关巨噬细胞亚群在TICs附近(即50μm半径内)积聚。这些先前未被重视的FcεRIα+巨噬细胞由骨髓来源细胞分化并替代性激活,通过IL-33–ST2–NF-κB途径创造了一个富含TGF-β的微环境,诱导对TICs的旁分泌TGF-β信号,并进一步上调IL-33表达。该途径的阻断或FcεRIα+巨噬细胞的耗竭减少了对TGF-β反应的TICs数量、侵袭性肿瘤进展率和化疗耐药性。
治疗耐药的TICs被认为是癌症治疗失败的主要元凶。通过研究小鼠模型,我们揭示了促进SCC恶性进展和耐药的TICs微环境形成的细胞和分子基础。TICs与FcεRIα+巨噬细胞之间IL-33–TGF-β微环境信号循环的发现为自我强化的TICs-微环境相互作用提供了机制性见解,这可能成为破坏TICs稳定性以提高癌症治疗效果的潜在靶点。
靶向肿瘤起始细胞(TICs)与微环境之间的相互作用是癌症治疗的一个有吸引力的途径。我们在此使用鳞状细胞癌小鼠模型证明,TICs在创建肿瘤进展和耐药所需的微环境中起着关键作用。TICs中由转录因子NRF2介导的抗氧化活性促进了核细胞因子白细胞介素-33(IL-33)的释放。这种细胞因子促进表达高亲和力免疫球蛋白E受体FcεRIα且邻近TICs的巨噬细胞分化。反过来,这些对IL-33反应的FcεRIα+巨噬细胞向TICs发送旁分泌转化生长因子β(TGF-β)信号,诱导侵袭性和耐药性,并进一步上调IL-33表达。这种由TICs驱动的IL-33–TGF-β前馈循环有可能被用于癌症治疗。
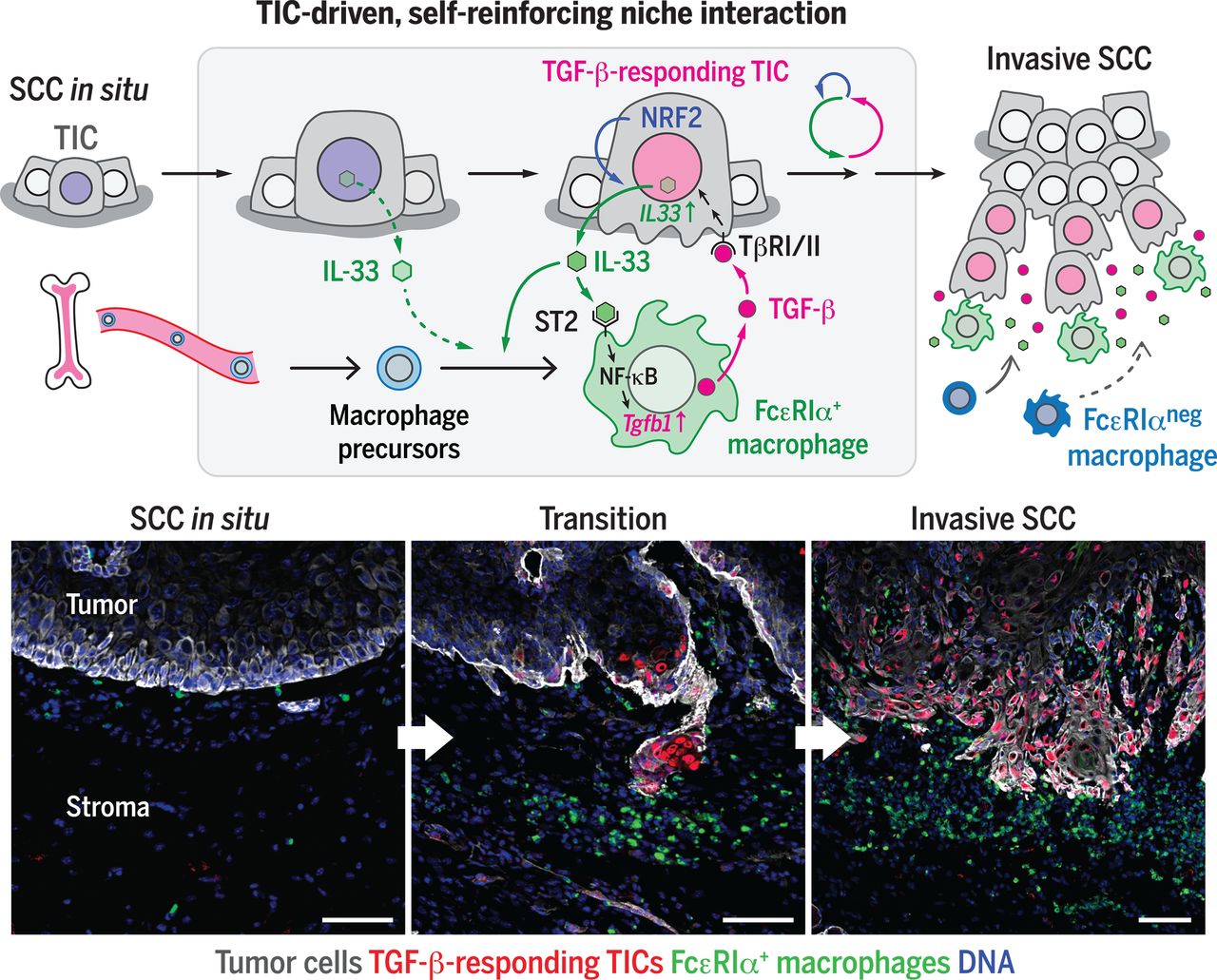
图:肿瘤起始细胞微环境中促进小鼠癌症进展的信号环路:响应TGF-β的肿瘤起始细胞通过NRF2介导的抗氧化反应释放IL-33,诱导未成熟髓系细胞分化为紧邻肿瘤起始细胞的FcεRIα+巨噬细胞。随后,FcεRIα+巨噬细胞通过旁分泌途径向肿瘤起始细胞传递TGF-β信号,促进鳞状细胞癌的侵袭进展及耐药性,并进一步诱导IL-33的释放,从而形成自我强化的信号环路。
Tumor-initiating cells establish an IL-33–TGF-β niche signaling loop to promote cancer progression